Strand NGS supports a workflow for the analysis and visualization of ChIP-Seq data. This workflow provides the ability to identify transcription factor binding sites and histone modification sites using either Enriched Region Detection, PICS, or MACS peak detection algorithms. It supports the ability to detect motifs in the peak regions using GADEM, and scan for known motifs in the genome or regions of interest. Normalized read coverage for the Transcription Start Sites can be viusalized using the TSS Plot. Further, downstream analysis such as GO, pathway analysis, etc. can be performed on the set of affected genes.
Download the ChIP-Seq Highlights Guide
White paper Calling narrow and broad peaks from ChIP-Seq data in StrandNGS
Watch the ChIP-Seq Webinar Recording
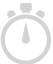
Identify protein binding sites using either Enriched Region Detection, PICS, or, MACS peak finding algorithms. View the results in the Genome Browser the context of known genes.
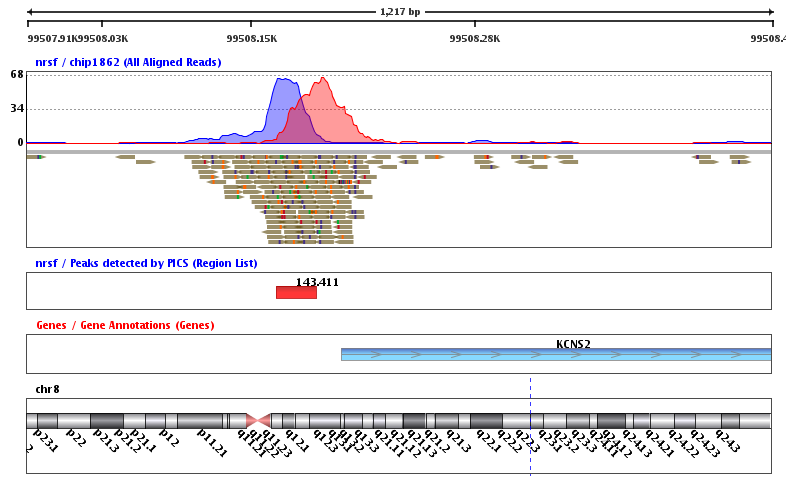
Discover the most significant motifs present in the vicinity of the binding regions using an efficient implementation of the GADEM algorithm. Motifs in JASPAR format can be imported and occurrences of the motifs can be detected by scanning the whole genome.
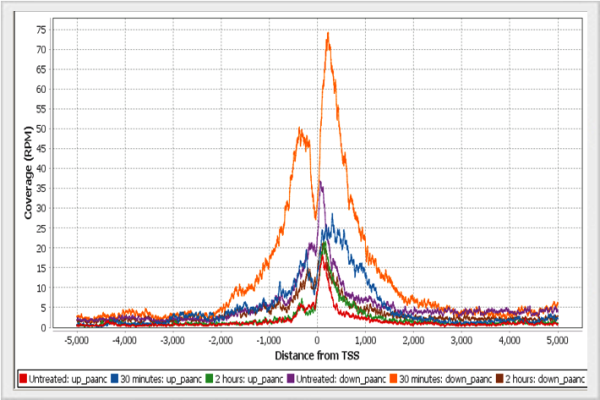
Visualize the profile of normalized reads around Transcription Start Sites.
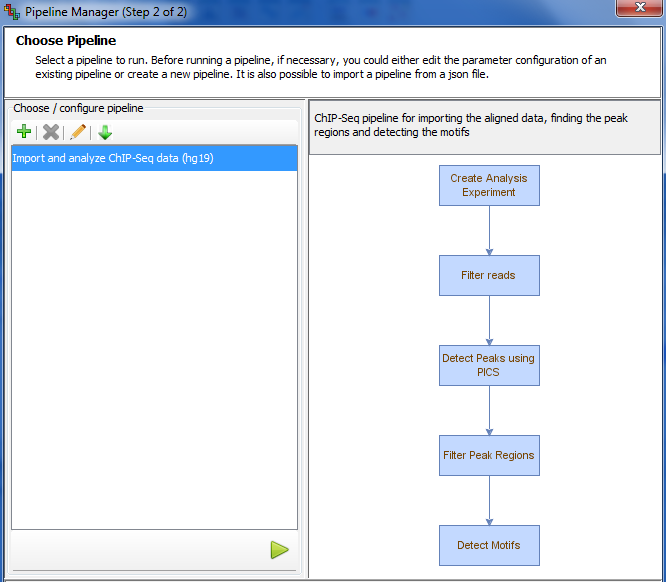
Execute one-shot pipeline for quick analysis and compute-intensive tasks using the Pipeline Manager. Also, configure pipelines or import customized pipelines using a .json file to perform reiterative tasks. Pipeline Manager also allows interaction with the user interface even as the pipelines are being executed in the background.
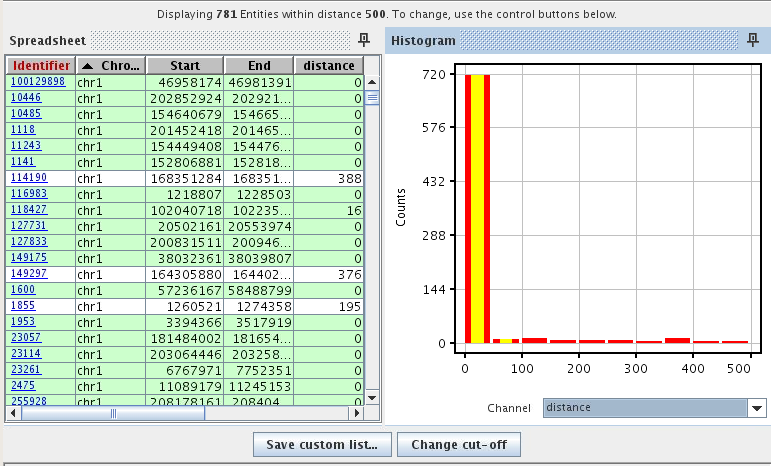
Identify genes affected by transcription factor regulation.
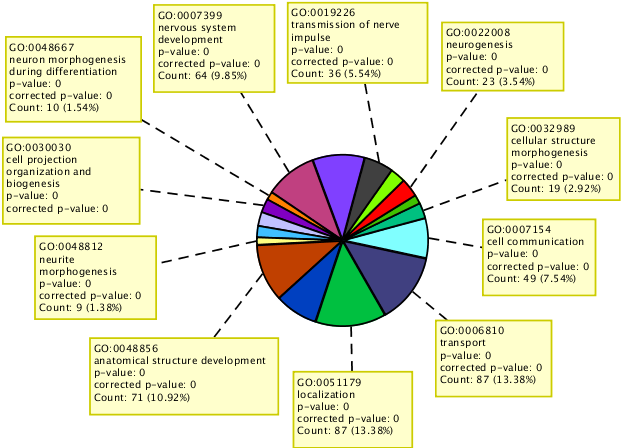
Perform GO analysis to identify significant GO terms for affected genes.
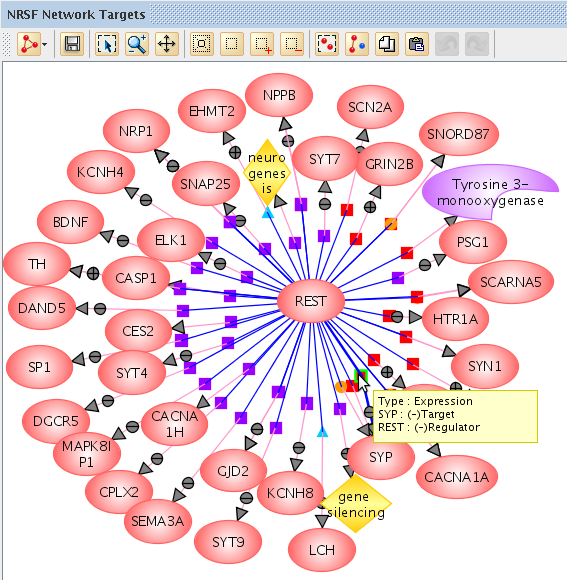
Use the packaged database of 2 million interactions (with supporting PubMed references) to find relationships between genes. Learn more
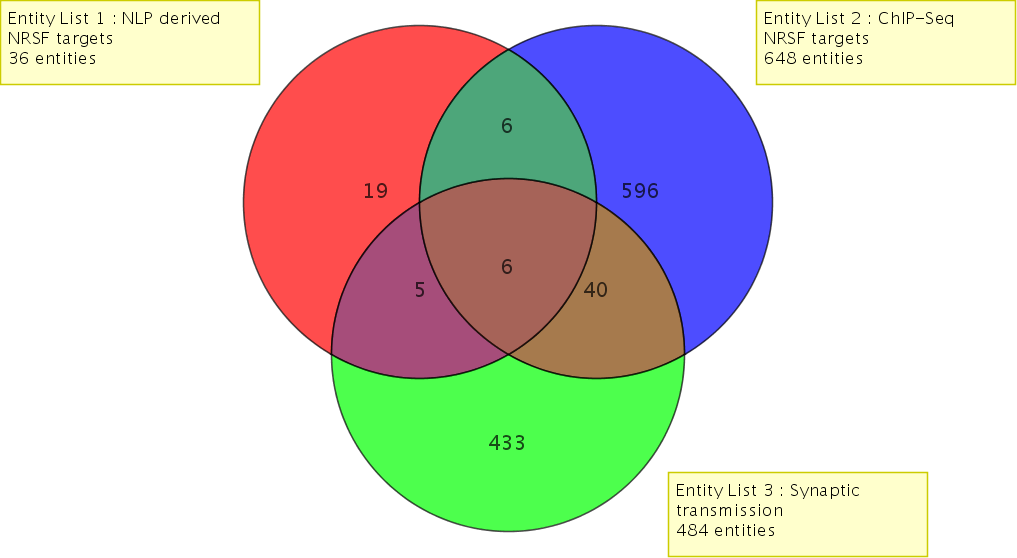
Compare different gene lists from multiple experiments and across organisms.
2018 © Strand Life Sciences Pvt Ltd. All rights reserved.