
Strand NGS v2.5 Release
The Strand NGS team is happy to announce the release of Strand NGS v2.5. This new version comes with exciting features like split read alignment, new workflow for MeDIP-Seq analysis, enhancements to facilitate an improved handling of samples from a large scale RNA-Seq study, a new SV detection algorithm using split reads and improved CNV visualizations. Here is a preview of some of these new features. Please see the release notes for full details.
MeDIP-Seq analysis
Strand NGS now has comprehensive support for methylation analysis. In addition to the already existing workflow for bisulfite sequencing, a new workflow for the analysis of MeDIP-Seq data has been introduced. This workflow includes calibration curve-based data normalization, CpG coverage analysis, methylation signal detection for regions of interest (ROIs) or whole genome, and detection of differentially methylated regions (DMR) to produce hypo- and hyper-methylated regions / genes.
View image on: Calibration Plot
Visualization of copy number variations
In targeted CNV workflow, CNV results can now be visualized in genome browser to simultaneously show copy number and z-scores for all the samples in the same track. This helps to quickly identify regions showing high confidence amplifications and deletions. These results can also be visualized in a web browser to show the copy number (CN) values along with z-scores in single- or multi-sample view across the whole-genome. Additional annotations such as GC%, overlapping genes, and cytoband information are also shown with detailed chromosome view.
View CNV visualizations on: Targeted CNV in Genome Browser ; Single Experiment in Web Browser ; Multiple Experiments in Web Browser
Split read alignment
Split read alignment is now possible when you perform DNA alignment in Strand NGS. In split read alignment, an input read splits into two segments, mapping each segment to a different location on the reference. The process of splitting is meant to detect fused reads, i.e., reads that specify a potential fusion between non-adjacent reference loci.
Structural variant detection using split reads
A new analysis step has been introduced to detect structural variants using split reads in DNA-Seq analysis workflow. Split read evidence is crucial to identify the locations of breakpoints for a variety of structural variants including large scale deletions, insertions, tandem duplications, inversions, translocations, and inverted translocations.
View image on: Structural Variants with Split Reads in Genome Browser
Correlation analysis
Perform a multi-omics correlation analysis of both miRNAs and mRNAs expression across your samples to extract negatively correlated miRNA and mRNA pairs and identify potential miRNA-targets.
View image on: Correlation Analysis
Enhancements to RNA-Seq workflow to handle large scale projects
The tool now comes with an efficient way to handle large-scale RNA-Seq projects where large number of samples arrive in batches over a long period. In such cases, separate experiments can be created for each batch of samples and quantification performed on them. These experiments can later be combined for an overall analysis without the need to run the compute-intensive quantification step again.
View image on: The Schematic Overview
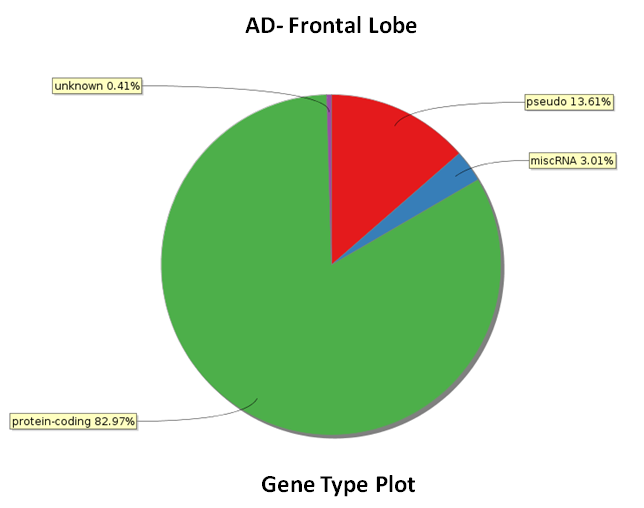
New post-alignment QC plots included in the RNA-Seq workflow
Post alignment QC has two new plots 'Genic Region QC Plot' and 'Gene Type Plot' in the Quality Control Manager of RNA-Seq workflow. Genic region QC plot displays the distribution of reads mapping to different regions of the transcriptome and genome, whereas the Gene type plot displays the distribution of the aligned reads mapping to different gene types.
View image on: Genic Region QC Plot and Gene Type Plot
Several other new features and enhancements are made in Strand NGS v2.5. All these new features are available once you update the product by clicking on Update Product from the Help menu.
We thank you for your continued support and invaluable feedback. For any queries, please feel free to contact us 24/5.
Email: sales@strandngs.com Phone (USA): 1-800-752-9122 Phone (Worldwide): +1-650-353-5060 |
Email: support@strandngs.com Phone (USA): 1-800-516-5181 Phone (Worldwide): +1-650-288-4559 |
Thank You
Strand NGS Team
 |
 |
 |
 |
 |
 |
 |
 |
 |









{kind=link}