Strand NGS supports an extensive workflow for the analysis and visualization of RNA-Seq data. The workflow includes standard differential expression analysis for different experimental conditions, as well as differential splicing analysis. It supports novel discovery including identifying novel genes and exons and novel splice junctions. It includes the ability to detect variants in the transcriptome, and the ability to detect gene fusion events. Further downstream analysis such as GO, pathway analysis, etc can be performed on the set of interesting genes.
Download the RNA-Seq Highlights Guide
Download the Large Scale Transcriptomics and Unique Molecular Identifiers Support
Watch the webinar recording on Unique Molecular Identifier-powered Ultra-sensitive Variant Calling
Watch the RNA-Seq Webinar Recording

Expression values at gene, exon and transcript level. RPKM, DESeq, Quantile and TMM methods for normalization.
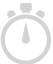
t-Test, Mann-Whitney, n-way ANOVA, and DESeq for identifying differentially expressed genes under different experimental conditions. Multiple testing correction using Bonferroni and Benjamini Hochberg methods.
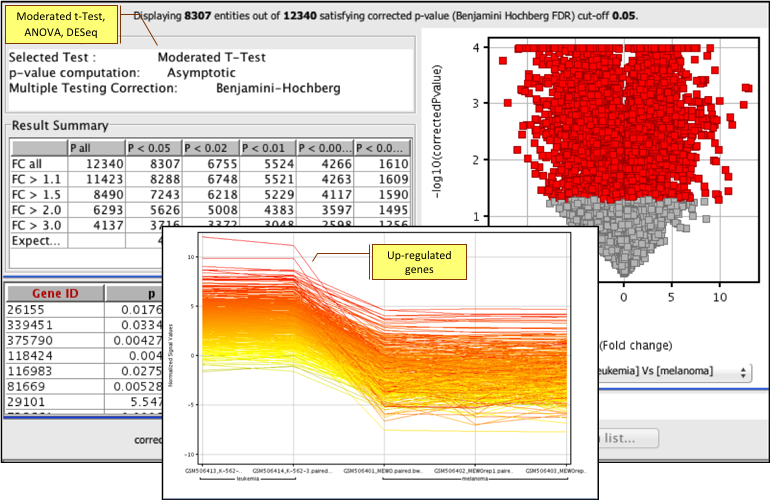
Determine gene and deconvoluted transcript expression profiles using EM algorithm; identify alternative splicing patterns. Learn more
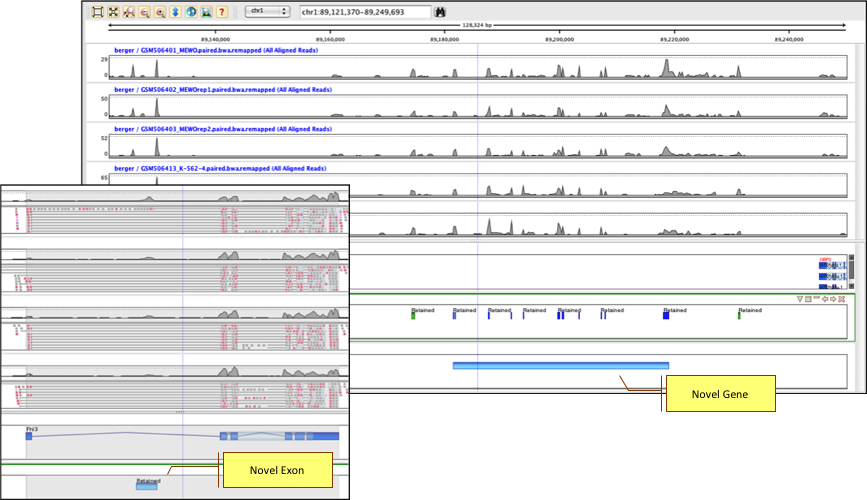
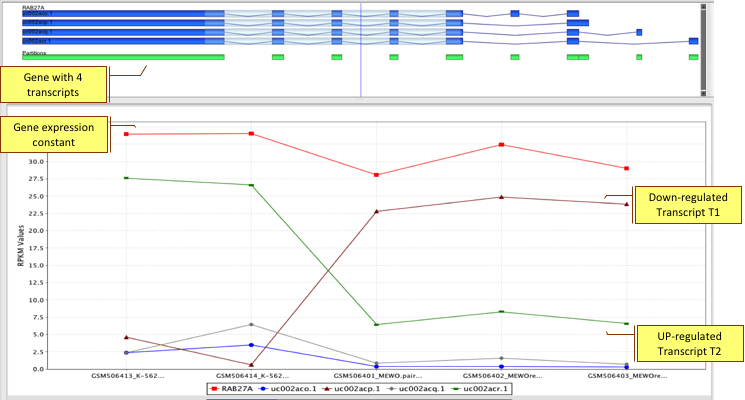
Analyze coverage patterns to detect novel genes and exons not present in NCBI, UCSC, and Ensembl annotations.
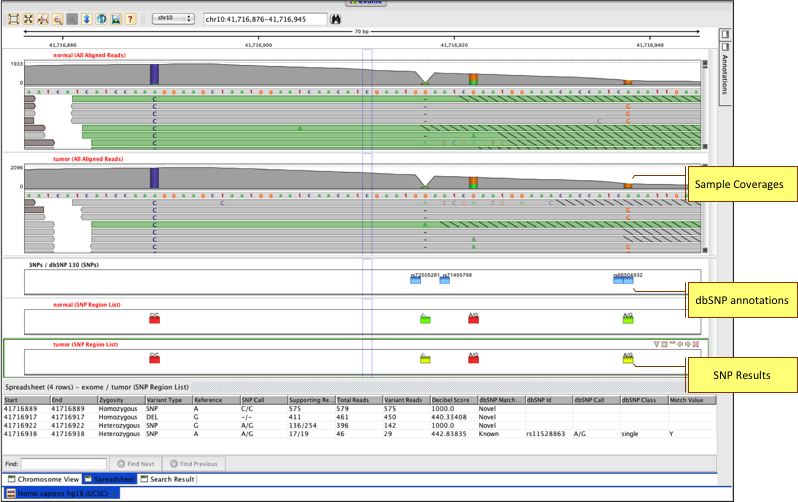
Identify variants in transcriptome, predict effect on transcripts, and perform differential SNP analysis.
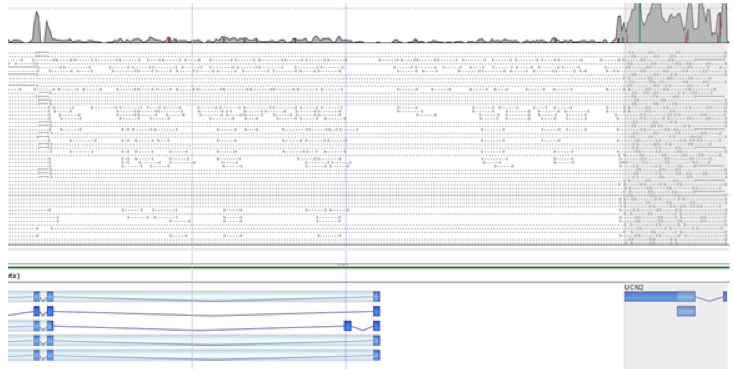
Find gene fusions via spliced and paired reads spanning the fused genes. Results annotated with paralogs and pseudogenes information for identifying false positives.
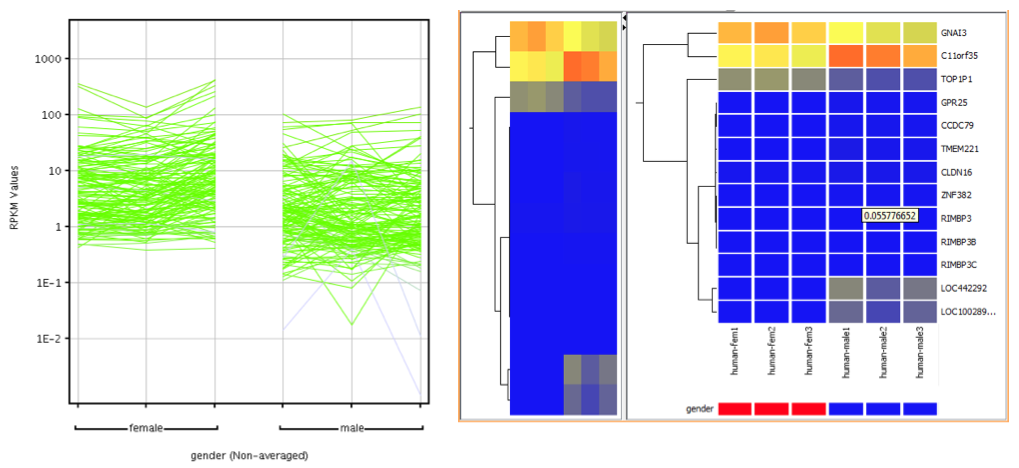
Cluster genes on their expression values to identify groups of genes behaving similarly across conditions.

Execute one-shot pipeline for quick analysis and compute-intensive tasks using the Pipeline Manager. Also, configure pipelines or import customized pipelines using a .json file to perform reiterative tasks. Pipeline Manager also allows interaction with the user interface even as the pipelines are being executed in the background.
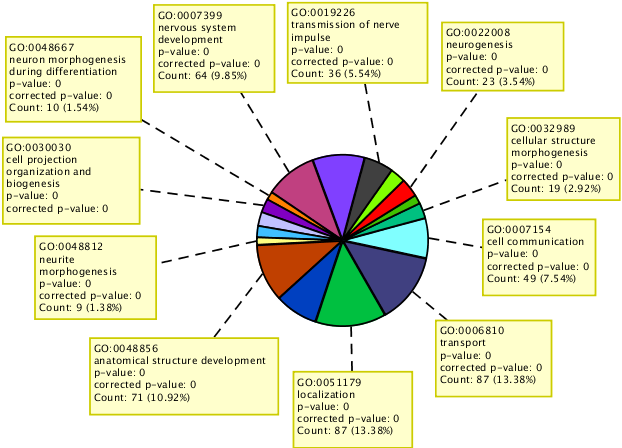
Perform GO analysis on identified set of interesting genes.
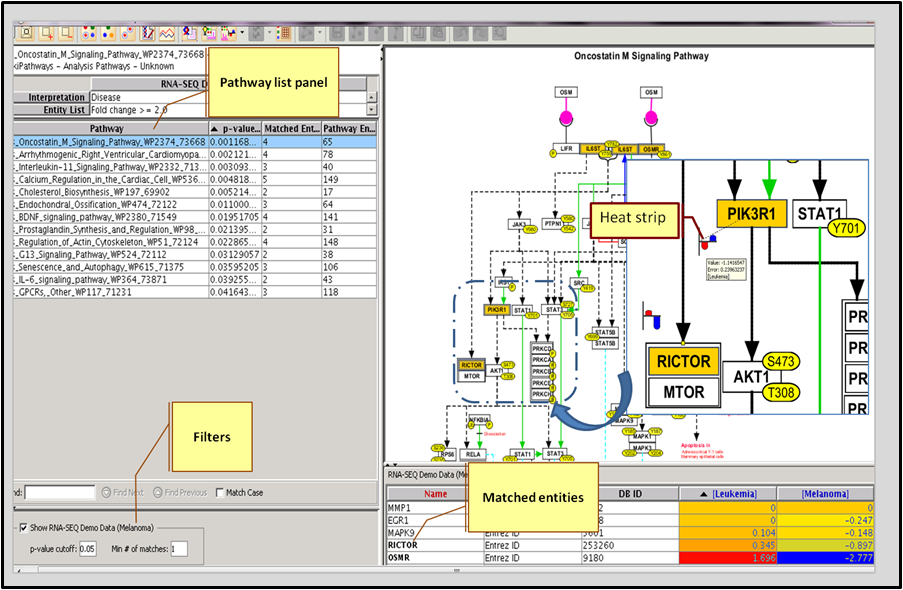
Use the packaged Interaction Database of over 2 million interactions (with supporting PubMed references) or other curated pathways to find relationships between genes. Learn more
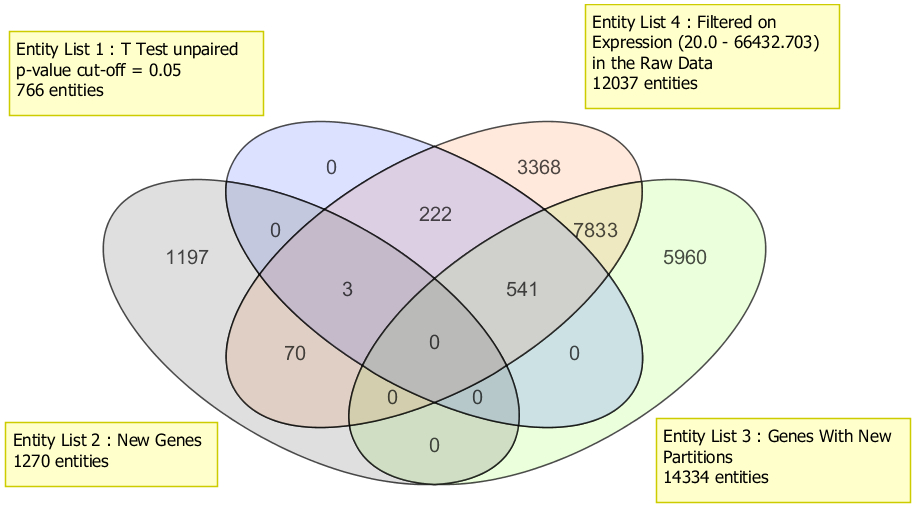
Compare different gene lists from multiple experiments and across organisms.
2018 © Strand Life Sciences Pvt Ltd. All rights reserved.