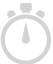
|
||||||||
|
|||||||||
";" string(19) "s:11:"Superfish 1";" string(24) "s:16:"menu-main-menu:0";" string(20) "s:12:"active-trail";" string(12) "s:5:"white";" string(796) "a:25:{s:11:"toggle_logo";i:1;s:11:"toggle_name";i:0;s:13:"toggle_slogan";i:0;s:14:"toggle_mission";i:1;s:24:"toggle_node_user_picture";i:0;s:27:"toggle_comment_user_picture";i:0;s:13:"toggle_search";i:0;s:14:"toggle_favicon";i:1;s:20:"toggle_primary_links";i:0;s:22:"toggle_secondary_links";i:0;s:12:"default_logo";i:0;s:9:"logo_path";s:38:"sites/default/files/sitetheme_logo.jpg";s:11:"logo_upload";s:0:"";s:15:"default_favicon";i:1;s:12:"favicon_path";s:0:"";s:14:"favicon_upload";s:0:"";s:17:"zen_block_editing";i:1;s:14:"zen_breadcrumb";s:3:"yes";s:24:"zen_breadcrumb_separator";s:5:" › ";s:19:"zen_breadcrumb_home";i:1;s:23:"zen_breadcrumb_trailing";i:1;s:20:"zen_breadcrumb_title";i:0;s:20:"zen_rebuild_registry";i:0;s:10:"zen_layout";s:18:"zen-columns-liquid";s:14:"zen_wireframes";i:0;}" string(9) "s:2:"-1";" string(18) "s:10:"horizontal";" string(272) "s:263:" ";" string(8) "s:1:"1";" string(26) "s:18:"Strand NGS License";" string(646) "s:637:"
We completed your order for Strand NGS software. Your license key or 'Order ID' information is below. (Please note: there may be multiple licenses as part of your order.)
[license-keys]
If Strand NGS is not installed, start with downloading the appropriate Strand NGS installer from http://strand-ngs.com/download and run the installer. Use the information above to activate the software. If you need help, please contact support via email at support@strandngs.com or go online to http://strand-ngs.com/contact/support. We hope you enjoy your experience using Strand NGS. Thank you, The Strand NGS Team
";" string(111) "s:102:"!company !first_name !last_name !street1 !street2 !city, !zone_code !postal_code !country_name_if";" string(99) "s:91:"!company !first_name !last_name !street1 !street2 !city, !postal_code !country_name_if";" string(111) "s:102:"!company !first_name !last_name !street1 !street2 !city, !zone_code !postal_code !country_name_if";" string(100) "s:92:"!company !first_name !last_name !street1 !street2 !postal_code !zone_name !country_name";" string(110) "s:101:"!company !first_name !last_name !street1 !street2 !city !zone_code !postal_code !country_name_if";" string(98) "s:90:"!company !first_name !last_name !street1 !street2 !city !postal_code !country_name_if";" string(110) "s:101:"!company !first_name !last_name !street1 !street2 !city !zone_code !postal_code !country_name_if";" string(110) "s:101:"!company !first_name !last_name !street1 !street2 !city !zone_code !postal_code !country_name_if";" string(110) "s:101:"!company !first_name !last_name !street1 !street2 !city !zone_code !postal_code !country_name_if";" string(110) "s:101:"!company !first_name !last_name !street1 !street2 !city !zone_code !postal_code !country_name_if";" string(93) "s:85:"!company !first_name !last_name !street1 !street2 !postal_code !city !country_name_if";" string(110) "s:101:"!company !first_name !last_name !street1 !street2 !city !zone_code !postal_code !country_name_if";" string(113) "s:104:"!company !first_name !last_name !street1 !street2 !city - !postal_code !zone_code !country_name_if";" string(110) "s:101:"!company !first_name !last_name !street1 !street2 !city !zone_code !postal_code !country_name_if";" string(110) "s:101:"!company !first_name !last_name !street1 !street2 !city !zone_code !postal_code !country_name_if";" string(109) "s:100:"!company !first_name !last_name !street1 !street2 !city, !postal_code !zone_name, !country_name";" string(110) "s:101:"!first_name !last_name !company !street1 !street2 !zone_name !postal_code !city !country_name_if";" string(110) "s:101:"!company !first_name !last_name !street1 !street2 !city !zone_code !postal_code !country_name_if";" string(109) "s:100:"!company !first_name !last_name !street1 !street2 !city, !postal_code !zone_name, !country_name";" string(111) "s:102:"!company !last_name !first_name !street1 !street2 !city, !zone_name !postal_code !country_name_if";" string(110) "s:101:"!company !first_name !last_name !street1 !street2 !city !zone_code !postal_code !country_name_if";" string(110) "s:101:"!company !first_name !last_name !street1 !street2 !city !zone_code !postal_code !country_name_if";" string(99) "s:91:"!company !first_name !last_name !street1 !street2 !postal_code !zone_name !country_name";" string(98) "s:90:"!company !first_name !last_name !street1 !street2 !postal_code !city !country_name_if";" string(98) "s:90:"!company !first_name !last_name !street1 !street2 !postal_code !city !country_name_if";" string(110) "s:101:"!company !first_name !last_name !street1 !street2 !city !zone_code !postal_code !country_name_if";" string(110) "s:101:"!company !first_name !last_name !street1 !street2 !city !zone_code !postal_code !country_name_if";" string(110) "s:101:"!company !first_name !last_name !street1 !street2 !city !zone_name !postal_code !country_name_if";" string(110) "s:101:"!company !first_name !last_name !street1 !street2 !city !zone_code !postal_code !country_name_if";" string(111) "s:102:"!company !first_name !last_name !street1 !street2 !postal_code !city !zone_name !country_name_if";" string(98) "s:90:"!company !first_name !last_name !street1 !street2 !postal_code !city !country_name_if";" string(93) "s:85:"!company !first_name !last_name !street1 !street2 !postal_code !city !country_name_if";" string(110) "s:101:"!company !first_name !last_name !street1 !street2 !city !zone_code !postal_code !country_name_if";" string(93) "s:85:"!company !first_name !last_name !street1 !street2 !postal_code !city !country_name_if";" string(110) "s:101:"!company !first_name !last_name !street1 !street2 !city !zone_code !postal_code !country_name_if";" string(99) "s:91:"!company !first_name !last_name !street1 !street2 !postal_code, !city !country_name_if";" string(98) "s:90:"!company !first_name !last_name !street1 !street2 !postal_code !city !country_name_if";" string(110) "s:101:"!company !first_name !last_name !street1 !street2 !city !zone_code !postal_code !country_name_if";" string(87) "s:79:"!company !first_name !last_name !street1 !street2 !zone_name, !country_name";" string(113) "s:104:"!company !first_name !last_name !street1 !street2 !city - !postal_code !zone_code !country_name_if";" string(94) "s:86:"!company !first_name !last_name !street1 !street2 !city !postal_code !country_name_if";" string(101) "s:93:"!first_name !last_name !country_name - !zone_name - !city, !street1 !street2 !postal_code";" string(111) "s:102:"!company !first_name !last_name !street1 !street2 !city, !zone_name !postal_code !country_name_if";" string(110) "s:101:"!company !first_name !last_name !street1 !street2 !city !zone_code !postal_code !country_name_if";" string(110) "s:101:"!company !first_name !last_name !street1 !street2 !city !zone_code !postal_code !country_name_if";" string(112) "s:103:"!company !first_name !last_name !street1 !street2 !postal_code, !city !zone_code !country_name_if";" string(101) "s:93:"!company !first_name !last_name !street1 !street2 !city - !postal_code !country_name_if";" string(110) "s:101:"!postal_code !zone_name!city !street1 !street2 !company !last_name !first_name !country_name_if";" string(93) "s:85:"!company !first_name !last_name !street1 !street2 !postal_code !city !country_name_if";" string(110) "s:101:"!company !first_name !last_name !street1 !street2 !city !zone_code !postal_code !country_name_if";" string(110) "s:101:"!company !first_name !last_name !street1 !street2 !city !zone_code !postal_code !country_name_if";" string(99) "s:91:"!company !first_name !last_name !street1 !street2 !city, !postal_code !country_name_if";" string(93) "s:85:"!company !first_name !last_name !street1 !street2 !postal_code !city !country_name_if";" string(110) "s:101:"!company !first_name !last_name !street1 !street2 !city !zone_code !postal_code !country_name_if";" string(110) "s:101:"!company !first_name !last_name !street1 !street2 !city !zone_code !postal_code !country_name_if";" string(110) "s:101:"!company !first_name !last_name !street1 !street2 !city !zone_code !postal_code !country_name_if";" string(111) "s:102:"!company !first_name !last_name !street1 !street2 !city, !zone_code !postal_code !country_name_if";" string(110) "s:101:"!company !first_name !last_name !street1 !street2 !city !zone_code !postal_code !country_name_if";" string(110) "s:101:"!company !first_name !last_name !street1 !street2 !city !zone_code !postal_code !country_name_if";" string(93) "s:85:"!company !first_name !last_name !street1 !street2 !postal_code !city !country_name_if";" string(110) "s:101:"!company !first_name !last_name !street1 !street2 !city !zone_code !postal_code !country_name_if";" string(110) "s:101:"!company !first_name !last_name !street1 !street2 !city !zone_code !postal_code !country_name_if";" string(110) "s:101:"!company !first_name !last_name !street1 !street2 !city !zone_code !postal_code !country_name_if";" string(94) "s:86:"!company !first_name !last_name !street1 !street2 !city, !zone_name !country_name";" string(110) "s:101:"!company !first_name !last_name !street1 !street2 !city !zone_code !postal_code !country_name_if";" string(110) "s:101:"!company !first_name !last_name !street1 !street2 !city !zone_code !postal_code !country_name_if";" string(93) "s:85:"!company !first_name !last_name !street1 !street2 !postal_code !city !country_name_if";" string(110) "s:101:"!company !first_name !last_name !street1 !street2 !city !zone_code !postal_code !country_name_if";" string(98) "s:90:"!first_name !last_name !company !street1 !street2 !city !postal_code !country_name_if";" string(110) "s:101:"!company !first_name !last_name !street1 !street2 !city !zone_code !postal_code !country_name_if";" string(113) "s:104:"!company !first_name !last_name !street1 !street2 !city - !postal_code !zone_code !country_name_if";" string(98) "s:90:"!company !first_name !last_name !street1 !street2 !postal_code !city !country_name_if";" string(97) "s:89:"!company !first_name !last_name !street1 !street2 !city !zone_name, !country_name_if";" string(110) "s:101:"!company !first_name !last_name !street1 !street2 !city !zone_code !postal_code !country_name_if";" string(110) "s:101:"!company !first_name !last_name !street1 !street2 !city !zone_name !postal_code !country_name_if";" string(111) "s:102:"!company !first_name !last_name !street1 !street2 !city, !zone_name !postal_code !country_name_if";" string(111) "s:102:"!company !first_name !last_name !street1 !street2 !city, !zone_code !postal_code !country_name_if";" string(94) "s:86:"!company !first_name !last_name !street1 !street2 !city, !zone_name !country_name";" string(110) "s:101:"!company !first_name !last_name !street1 !street2 !city !zone_code !postal_code !country_name_if";" string(110) "s:101:"!company !first_name !last_name !street1 !street2 !city !zone_code !postal_code !country_name_if";" string(112) "s:103:"!company !first_name !last_name !street1 !street2 !city, !postal_code !zone_name, !country_name_if";" string(93) "s:85:"!company !first_name !last_name !street1 !street2 !postal_code !city !country_name_if";" string(110) "s:101:"!company !first_name !last_name !street1 !street2 !city !zone_code !postal_code !country_name_if";" string(99) "s:91:"!company !first_name !last_name !street1 !street2 !city, !postal_code !country_name_if";" string(110) "s:101:"!company !first_name !last_name !street1 !street2 !city !zone_code !postal_code !country_name_if";" string(110) "s:101:"!company !first_name !last_name !street1 !street2 !city !zone_code !postal_code !country_name_if";" string(110) "s:101:"!company !first_name !last_name !street1 !street2 !city !zone_code !postal_code !country_name_if";" string(89) "s:81:"!company !first_name !last_name !street1 !street2 !country_name, !postal_code";" string(98) "s:90:"!company !first_name !last_name !street1 !street2 !postal_code !city !country_name_if";" string(110) "s:101:"!company !first_name !last_name !street1 !street2 !city !zone_code !postal_code !country_name_if";" string(99) "s:91:"!company !first_name !last_name !street1 !street2 !city !postal_code !country_name_if";" string(93) "s:85:"!company !first_name !last_name !street1 !street2 !postal_code !city !country_name_if";" string(110) "s:101:"!company !first_name !last_name !street1 !street2 !city !zone_code !postal_code !country_name_if";" string(110) "s:101:"!company !first_name !last_name !street1 !street2 !city !zone_code !postal_code !country_name_if";" string(98) "s:90:"!company !first_name !last_name !street1 !street2 !postal_code !city !country_name_if";" string(98) "s:90:"!company !first_name !last_name !street1 !street2 !postal_code !city !country_name_if";" string(111) "s:102:"!company !last_name !first_name !street1 !street2 !city, !zone_name !postal_code !country_name_if";" string(110) "s:101:"!company !first_name !last_name !street1 !street2 !city !zone_code !postal_code !country_name_if";" string(107) "s:99:"!first_name !last_name !company !street1 !street2 !city, !zone_name !postal_code !country_name";" string(110) "s:101:"!company !first_name !last_name !street1 !street2 !city !zone_code !postal_code !country_name_if";" string(101) "s:93:"!company !first_name !last_name !street1 !street2 !postal_code, !city !country_name_if";" string(101) "s:93:"!country_name !zone_name !city !postal_code !street1 !street2 !company !first_name !last_name";" string(97) "s:89:"!company !first_name !last_name !street1 !street2 !city, !zone_code !country_name_if";" string(99) "s:91:"!first_name !last_name !company !street1 !street2 !city !postal_code !country_name_if";" string(110) "s:101:"!company !first_name !last_name !street1 !street2 !city !zone_code !postal_code !country_name_if";" string(110) "s:101:"!company !first_name !last_name !street1 !street2 !city !zone_code !postal_code !country_name_if";" string(111) "s:102:"!company !first_name !last_name !street1 !street2 !city, !zone_code !postal_code !country_name_if";" string(110) "s:101:"!company !first_name !last_name !street1 !street2 !city !zone_code !postal_code !country_name_if";" string(110) "s:101:"!company !first_name !last_name !street1 !street2 !city !zone_code !postal_code !country_name_if";" string(4) "i:0;" string(4) "i:0;" string(4) "i:1;" string(4) "i:0;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(7) "s:0:"";" string(4) "i:1;" string(8) "s:1:"0";" string(13) "s:6:"Signup";" string(42) "s:34:"Signups are closed for this event.";" string(4) "i:1;" string(15) "s:8:"thickbox";" string(4) "i:1;" string(4) "i:1;" string(19) "s:11:"Add to cart";" string(14) "s:7:"Buy Now";" string(8) "s:1:"1";" string(7) "s:0:"";" string(60) "s:52:"An administrator created an account for you at !site";" string(477) "s:468:"!username, A site administrator at !site has created an account for you. You may now log in to !login_uri using the following username and password: username: !username password: !password You may also log in by clicking on this link or copying and pasting it in your browser: !login_url This is a one-time login, so it can be used only once. After logging in, you will be redirected to !edit_uri so you can change your password. -- !site team";" string(25) "s:17:"Welcome to !site ";" string(6270) "s:6260:"
|
|||||||||
|
|||||||||
Hi [profile-name], We received your payment for Strand NGS. Thanks! Before we send you your license information, we need to verify that you are currently a GeneSpring user. This will take up to 2 business days. After we verify your account, we will send your Strand NGS license information. If you have any questions, you can contact our Support team here (http://strand-ngs.com/contact/support). Thanks for your patience! The Strand NGS Team
";" string(8) "s:1:"1";" string(7) "s:0:"";" string(8) "s:1:"1";" string(274) "s:265:"The purchase process is almost complete. Review the details below and click 'Submit Order' if all the information is correct. You will be taken to a PayPal site to securely make your payment. Please remember that you do NOT need a PayPal account to make the payment";" string(8) "s:1:"1";" string(36) "s:28:"Thank you for your business!";" string(8) "s:1:"1";" string(102) "s:94:"We will email your PayPal payment confirmation and Avadis NGS license information to you soon.";" string(8) "s:1:"1";" string(8) "s:1:"0";" string(8) "s:1:"1";" string(4) "i:0;" string(8) "s:1:"1";" string(8) "s:1:"0";" string(12) "s:5:"86400";" string(8) "s:1:"0";" string(41) "a:2:{i:0;s:6:"status";i:1;s:7:"promote";}" string(8) "s:1:"1";" string(45) "s:37:"form-3e27b6e4413857310dd9c78f419e6814";" string(15) "s:8:"disabled";" string(11) "s:4:"1000";" string(8) "s:1:"0";" string(11) "s:4:"none";" string(232) "a:9:{s:5:"title";s:2:"-5";s:10:"body_field";s:2:"-2";s:20:"revision_information";s:2:"-1";s:6:"author";s:1:"0";s:7:"options";s:1:"1";s:16:"comment_settings";s:1:"4";s:4:"menu";s:2:"-4";s:4:"path";s:1:"3";s:11:"attachments";s:1:"2";}" string(30) "s:22:"siteadmin@strandls.com";" string(27) "s:19:"The GeneSpring Team";" string(36) "s:28:"Form submission from: %title";" string(4) "i:0;" string(11) "s:4:"long";" string(16) "s:9:"delimited";" string(9) "s:2:"\t";" string(8) "s:1:"1";" string(6) "a:0:{}" string(41) "a:2:{i:0;s:6:"status";i:1;s:7:"promote";}" string(8) "s:1:"1";" string(45) "s:37:"form-86a07d7988dff727ffa41ecf46c02fed";" string(9) "s:2:"50";" string(8) "s:1:"3";" string(4) "i:0;" string(8) "s:1:"1";" string(8) "s:1:"1";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(11) "s:4:"none";" string(8) "s:1:"0";" string(7) "s:0:"";" string(4) "i:0;" string(8) "s:1:"0";" string(155) "a:1:{s:15:"field_cust_name";a:5:{s:3:"gid";s:14:"nodeaccess_uid";s:7:"enabled";i:0;s:10:"grant_view";i:0;s:12:"grant_update";i:0;s:12:"grant_delete";i:0;}}" string(4) "i:1;" string(8) "s:1:"0";" string(8) "s:1:"2";" string(4) "i:1;" string(6) "a:0:{}" string(7) "s:0:"";" string(4) "i:0;" string(8) "s:1:"2";" string(8) "s:1:"4";" string(8) "s:1:"1";" string(9) "s:2:"50";" string(8) "s:1:"3";" string(4) "i:0;" string(8) "s:1:"1";" string(8) "s:1:"1";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(11) "s:4:"none";" string(8) "s:1:"0";" string(7) "s:0:"";" string(4) "i:0;" string(21) "s:13:"UA-18926606-1";" string(8) "s:1:"0";" string(155) "a:16:{i:1;i:1;i:2;i:2;i:14;i:14;i:13;i:13;i:1000;i:0;i:1002;i:0;i:16;i:0;i:18;i:0;i:17;i:0;i:9;i:0;i:1003;i:0;i:6;i:0;i:15;i:0;i:7;i:0;i:3;i:0;i:1001;i:0;}" string(533) "a:12:{s:5:"roles";s:5:"roles";s:12:"profile_name";s:12:"profile_name";s:13:"profile_email";s:13:"profile_email";s:15:"profile_company";s:15:"profile_company";s:15:"profile_country";s:15:"profile_country";s:12:"profile_city";s:12:"profile_city";s:13:"profile_phone";s:13:"profile_phone";s:19:"profile_experiments";s:19:"profile_experiments";s:18:"profile_data_files";s:18:"profile_data_files";s:19:"profile_gx_customer";s:19:"profile_gx_customer";s:14:"profile_source";s:14:"profile_source";s:13:"profile_other";s:13:"profile_other";}" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(132) "s:123:"7z|aac|avi|csv|doc|exe|flv|gif|gz|jpe?g|js|mp(3|4|e?g)|mov|pdf|phps|png|ppt|rar|sit|tar|torrent|txt|wma|wmv|xls|xml|zip|bin";" string(4) "i:1;" string(4) "i:1;" string(4) "i:0;" string(52) "s:44:"_gaq.push(['_setAccount', 'UA-18926606-1']);";" string(7) "s:0:"";" string(13) "s:6:"footer";" string(8) "s:1:"1";" string(12) "s:5:"small";" string(11) "s:4:"node";" string(8) "s:1:"1";" string(18) "s:10:"embed-view";" string(18) "s:10:"embed-view";" string(32) "s:24:"signup_user_list:default";" string(38) "s:30:"signup_user_admin_list:default";" string(228) "a:8:{s:24:"signup_available_signups";b:1;s:22:"signup_current_signups";b:1;s:22:"signup_user_admin_list";b:1;s:16:"signup_user_list";b:1;s:11:"uc_products";b:1;s:12:"date_browser";b:1;s:7:"archive";b:0;s:13:"taxonomy_term";b:1;}" string(40) "s:32:"Avadis NGS Trial License Request";" string(514) "s:505:"Thank you for registering to evaluate Avadis NGS! All free trial requests are currently being queued up, and will be emailed to you by our support team before October 11, 2010. Free trial license requests and online purchases after October 11 will be delivered and fulfilled in real-time. If you have any questions, please contact support via email at avadisngs.support@strandsi.com or go online to http://avadis-ngs.com/support. We appreciate your patience, The Avadis NGS Team
";" string(540) "a:22:{s:8:"keywords";s:8:"keywords";s:10:"page_title";s:10:"page_title";s:8:"abstract";i:0;s:9:"canonical";i:0;s:9:"copyright";i:0;s:11:"description";i:0;s:19:"dcterms.contributor";i:0;s:15:"dcterms.creator";i:0;s:12:"dcterms.date";i:0;s:19:"dcterms.description";i:0;s:17:"dcterms.publisher";i:0;s:13:"dcterms.title";i:0;s:8:"location";i:0;s:13:"geo.placename";i:0;s:10:"geo.region";i:0;s:4:"logo";i:0;s:15:"original-source";i:0;s:13:"revisit-after";i:0;s:6:"robots";i:0;s:8:"shorturl";i:0;s:8:"standout";i:0;s:18:"syndication-source";i:0;}" string(36) "s:28:"files/images/icon-search.jpg";" string(4) "i:0;" string(35) "s:27:"Containing any of the words";" string(4) "i:0;" string(6) "a:0:{}" string(29) "s:21:"Containing the phrase";" string(4) "i:0;" string(4) "i:0;" string(4) "i:1;" string(10) "s:3:"350";" string(8) "s:1:"0";" string(8) "s:1:"3";" string(4) "i:1;" string(52) "a:2:{s:12:"imagebrowser";i:0;s:10:"img_assist";i:0;}" string(4) "i:1;" string(54) "a:5:{i:8;i:0;i:1001;i:0;i:16;i:0;i:1000;i:0;i:14;i:0;}" string(7) "s:0:"";" string(7) "s:0:"";" string(149) "a:8:{s:5:"index";i:0;s:7:"noindex";i:0;s:6:"follow";i:0;s:8:"nofollow";i:0;s:9:"noarchive";i:0;s:5:"noodp";i:0;s:9:"nosnippet";i:0;s:6:"noydir";i:0;}" string(13) "s:6:"Search";" string(4) "i:0;" string(14) "s:7:"private";" string(138) "s:129:"Hi [profile-name], We received your payment for Strand NGS. Thank you! You opted for Academic/Non-profit discount - please allow us to complete our verification of your non-profit credentials. This process usually takes less than 1 business day. We may get in touch with you in this regard. Meanwhile, please proceed to download the software from http://strand-ngs.com/download. If you have any questions, you can contact our Support team here (http://strand-ngs.com/support/contact-us). Thanks for your patience! The Strand NGS Team";" string(8) "s:1:"1";" string(26) "s:18:"Strand NGS License";" string(2) "N;" string(7) "s:0:"";" string(10) "s:3:"Log";" string(6) "a:0:{}" string(11) "s:4:"type";" string(11) "s:4:"each";" string(6) "a:0:{}" string(4) "i:0;" string(4) "i:0;" string(7) "s:0:"";" string(8) "s:1:"2";" string(8) "s:1:"4";" string(23) "a:1:{i:0;s:6:"enable";}" string(8) "s:1:"1";" string(8) "s:1:"1";" string(55) "s:47:" ";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(4) "i:1;" string(4) "i:0;" string(4) "i:0;" string(4) "i:0;" string(4) "i:0;" string(4) "i:0;" string(4) "i:0;" string(4) "i:0;" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(4) "b:1;" string(21) "a:1:{i:0;s:4:"book";}" string(11) "s:4:"book";" string(4) "i:0;" string(4) "i:0;" string(4) "i:0;" string(4) "i:0;" string(4) "i:0;" string(4) "i:0;" string(4) "i:0;" string(8) "s:1:"3";" string(8) "s:1:"3";" string(8) "s:1:"3";" string(8) "s:1:"3";" string(8) "s:1:"3";" string(8) "s:1:"3";" string(8) "s:1:"3";" string(8) "s:1:"4";" string(8) "s:1:"4";" string(8) "s:1:"4";" string(8) "s:1:"4";" string(8) "s:1:"4";" string(8) "s:1:"4";" string(8) "s:1:"4";" string(8) "s:1:"1";" string(8) "s:1:"1";" string(8) "s:1:"1";" string(8) "s:1:"1";" string(8) "s:1:"1";" string(8) "s:1:"2";" string(8) "s:1:"1";" string(9) "s:2:"50";" string(9) "s:2:"50";" string(9) "s:2:"50";" string(9) "s:2:"50";" string(9) "s:2:"50";" string(9) "s:2:"50";" string(9) "s:2:"50";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"1";" string(8) "s:1:"0";" string(4) "i:0;" string(8) "s:1:"1";" string(8) "s:1:"1";" string(8) "s:1:"1";" string(8) "s:1:"1";" string(8) "s:1:"1";" string(8) "s:1:"0";" string(8) "s:1:"1";" string(8) "s:1:"2";" string(8) "s:1:"2";" string(8) "s:1:"1";" string(8) "s:1:"1";" string(8) "s:1:"1";" string(8) "s:1:"1";" string(8) "s:1:"1";" string(8) "s:1:"0";" string(8) "s:1:"1";" string(8) "s:1:"0";" string(8) "s:1:"2";" string(4) "i:0;" string(8) "s:1:"0";" string(11) "s:4:"none";" string(11) "s:4:"none";" string(11) "s:4:"none";" string(11) "s:4:"none";" string(11) "s:4:"none";" string(11) "s:4:"none";" string(11) "s:4:"none";" string(11) "s:4:"none";" string(11) "s:4:"none";" string(11) "s:4:"none";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"1";" string(8) "s:1:"0";" string(270) "s:261:" ";" string(270) "s:261:" ";" string(334) "a:13:{s:5:"title";s:2:"-5";s:20:"revision_information";s:2:"14";s:6:"author";s:2:"15";s:7:"options";s:2:"16";s:16:"comment_settings";s:2:"19";s:4:"menu";s:2:"11";s:4:"path";s:2:"18";s:11:"attachments";s:2:"17";s:6:"domain";s:2:"12";s:4:"flag";s:1:"4";s:13:"path_redirect";s:2:"21";s:10:"xmlsitemap";s:2:"20";s:9:"nodewords";s:2:"13";}" string(304) "a:12:{s:5:"title";s:2:"-5";s:10:"body_field";s:2:"-2";s:20:"revision_information";s:1:"1";s:6:"author";s:1:"2";s:7:"options";s:1:"3";s:16:"comment_settings";s:1:"6";s:4:"menu";s:2:"-3";s:4:"path";s:1:"7";s:11:"attachments";s:1:"4";s:6:"domain";s:2:"-1";s:10:"xmlsitemap";s:1:"5";s:9:"nodewords";s:1:"0";}" string(304) "a:12:{s:5:"title";s:2:"-5";s:10:"body_field";s:2:"-2";s:20:"revision_information";s:1:"1";s:6:"author";s:1:"2";s:7:"options";s:1:"3";s:16:"comment_settings";s:1:"6";s:4:"menu";s:2:"-3";s:4:"path";s:1:"7";s:11:"attachments";s:1:"4";s:6:"domain";s:2:"-1";s:10:"xmlsitemap";s:1:"5";s:9:"nodewords";s:1:"0";}" string(310) "a:12:{s:5:"title";s:2:"-5";s:10:"body_field";s:1:"0";s:20:"revision_information";s:2:"20";s:6:"author";s:2:"20";s:7:"options";s:2:"25";s:16:"comment_settings";s:2:"30";s:4:"menu";s:2:"-2";s:4:"path";s:2:"30";s:11:"attachments";s:2:"30";s:6:"domain";s:1:"1";s:10:"xmlsitemap";s:2:"30";s:9:"nodewords";s:2:"10";}" string(339) "a:13:{s:5:"title";s:2:"34";s:20:"revision_information";s:2:"52";s:6:"author";s:2:"53";s:7:"options";s:2:"54";s:16:"comment_settings";s:2:"56";s:4:"menu";s:2:"49";s:8:"taxonomy";s:2:"39";s:4:"path";s:2:"55";s:11:"attachments";s:2:"60";s:6:"domain";s:2:"50";s:13:"path_redirect";s:2:"30";s:10:"xmlsitemap";s:2:"58";s:9:"nodewords";s:2:"51";}" string(4) "i:0;" string(4) "i:0;" string(4) "i:0;" string(4) "i:0;" string(4) "i:0;" string(4) "i:0;" string(4) "i:0;" string(4) "i:0;" string(23) "a:1:{i:0;s:6:"enable";}" string(4) "i:1;" string(198) "a:6:{s:7:"alchemy";s:7:"alchemy";s:3:"seo";s:3:"seo";s:10:"kwresearch";s:10:"kwresearch";s:11:"readability";s:11:"readability";s:9:"scribeseo";s:9:"scribeseo";s:11:"w3canalyzer";s:11:"w3canalyzer";}" string(7) "s:0:"";" string(23) "a:1:{i:0;s:6:"enable";}" string(4) "i:0;" string(4) "i:1;" string(29) "s:21:"
user/*/track
";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(9) "s:2:"25";" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(113) "s:104:"node/%n node/%n/edit comment/reply/%n node/add/book/parent/%n book/export/html/%n node/%n/outline
";" string(8) "s:1:"1";" string(440) "a:10:{i:0;a:4:{i:-1;i:1;i:2;i:1;i:3;i:1;i:4;i:1;}i:1;a:4:{i:-1;i:0;i:2;i:0;i:3;i:0;i:4;i:0;}i:2;a:4:{i:-1;i:1;i:2;i:1;i:3;i:1;i:4;i:1;}i:3;a:4:{i:-1;i:0;i:2;i:0;i:3;i:0;i:4;i:0;}i:6;a:4:{i:-1;i:0;i:2;i:0;i:3;i:0;i:4;i:0;}i:7;a:4:{i:-1;i:0;i:2;i:0;i:3;i:0;i:4;i:0;}i:9;a:4:{i:-1;i:0;i:2;i:0;i:3;i:0;i:4;i:0;}i:13;a:4:{i:-1;i:0;i:2;i:0;i:3;i:0;i:4;i:0;}i:14;a:4:{i:-1;i:0;i:2;i:0;i:3;i:0;i:4;i:0;}i:15;a:4:{i:-1;i:0;i:2;i:0;i:3;i:0;i:4;i:0;}}" string(21) "s:13:"174.129.43.92";" string(12) "s:5:"https";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(463) "a:1:{s:5:"slots";a:5:{i:1;a:4:{s:4:"slot";i:1;s:4:"name";s:10:"User roles";s:5:"value";s:17:"[user-role-names]";s:5:"scope";s:1:"1";}i:2;a:4:{s:4:"slot";i:2;s:4:"name";s:0:"";s:5:"value";s:0:"";s:5:"scope";s:1:"3";}i:3;a:4:{s:4:"slot";i:3;s:4:"name";s:0:"";s:5:"value";s:0:"";s:5:"scope";s:1:"3";}i:4;a:4:{s:4:"slot";i:4;s:4:"name";s:0:"";s:5:"value";s:0:"";s:5:"scope";s:1:"3";}i:5;a:4:{s:4:"slot";i:5;s:4:"name";s:0:"";s:5:"value";s:0:"";s:5:"scope";s:1:"3";}}}" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(26) "s:18:"Strand NGS Primary";" string(9) "s:2:"id";" string(8) "s:1:"0";" string(8) "s:1:"3";" string(57) "s:49:"taxonomy/term/%t taxonomy/edit/term/%t forum/%t";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(4) "i:0;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(9) "s:2:"90";" string(27) "s:19:"sites/default/files";" string(8) "s:1:"1";" string(7) "i:6103;" string(4) "i:0;" string(4) "i:0;" string(4) "i:0;" string(45) "s:37:"form-30e381d395cd12c710268c0c37ce83dd";" string(45) "s:37:"form-69b869dbf0e61da05310fae3cdc45c40";" string(45) "s:37:"form-3d95dd8c4be3b11e02edc6cd9174394f";" string(45) "s:37:"form-8ae9d4d155184a8da7d98d77dfcf2c66";" string(45) "s:37:"form-2ad298fb5f3b34d3547cc75ba5f74aa7";" string(45) "s:37:"form-b42e87d219f8712a0bf851ee1ce217da";" string(45) "s:37:"form-e3cb0409d112bc975ce02100e8871673";" string(45) "s:37:"form-82f6e4d9dd1baaa01120294669122f8c";" string(45) "s:37:"form-f5572751f38edb7531c2df7cfd069e60";" string(45) "s:37:"form-8190e689709795f6a32fa956edc38f7e";" string(7) "s:0:"";" string(52) "s:44:"_gaq.push(['_setAccount', 'UA-18926606-1']);";" string(557) "a:9:{s:9:"config_id";s:32:"content-taxonomy-field_hierarchy";s:12:"save_lineage";s:1:"1";s:15:"enforce_deepest";s:1:"0";s:12:"entity_count";s:1:"0";s:14:"require_entity";s:1:"0";s:9:"resizable";s:1:"1";s:12:"level_labels";a:2:{s:6:"status";i:0;s:6:"labels";a:1:{i:0;s:0:"";}}s:7:"dropbox";a:4:{s:6:"status";i:0;s:5:"title";s:14:"All selections";s:5:"limit";s:1:"0";s:8:"reset_hs";s:1:"1";}s:11:"editability";a:5:{s:6:"status";i:0;s:10:"item_types";a:1:{i:0;s:0:"";}s:14:"allowed_levels";a:1:{i:0;i:1;}s:16:"allow_new_levels";i:0;s:10:"max_levels";s:1:"0";}}" string(587) "a:9:{s:9:"config_id";s:11:"taxonomy-14";s:12:"save_lineage";s:1:"1";s:15:"enforce_deepest";s:1:"0";s:12:"entity_count";s:1:"0";s:14:"require_entity";s:1:"0";s:9:"resizable";s:1:"1";s:12:"level_labels";a:2:{s:6:"status";i:0;s:6:"labels";a:2:{i:0;s:8:"Category";i:1;s:12:"Sub-category";}}s:7:"dropbox";a:4:{s:6:"status";i:0;s:5:"title";s:14:"All selections";s:5:"limit";s:1:"0";s:8:"reset_hs";s:1:"1";}s:11:"editability";a:5:{s:6:"status";i:0;s:10:"item_types";a:2:{i:0;s:0:"";i:1;s:0:"";}s:14:"allowed_levels";a:2:{i:0;i:1;i:1;i:1;}s:16:"allow_new_levels";i:0;s:10:"max_levels";s:1:"1";}}" string(624) "a:9:{s:9:"config_id";s:11:"taxonomy-16";s:12:"save_lineage";s:1:"1";s:15:"enforce_deepest";s:1:"0";s:12:"entity_count";s:1:"0";s:14:"require_entity";s:1:"0";s:9:"resizable";s:1:"0";s:12:"level_labels";a:2:{s:6:"status";i:1;s:6:"labels";a:3:{i:0;s:7:"Product";i:1;s:8:"Category";i:2;s:12:"Sub-category";}}s:7:"dropbox";a:4:{s:6:"status";i:0;s:5:"title";s:14:"All selections";s:5:"limit";s:1:"0";s:8:"reset_hs";s:1:"1";}s:11:"editability";a:5:{s:6:"status";i:0;s:10:"item_types";a:3:{i:0;s:0:"";i:1;s:0:"";i:2;s:0:"";}s:14:"allowed_levels";a:3:{i:0;i:1;i:1;i:1;i:2;i:1;}s:16:"allow_new_levels";i:0;s:10:"max_levels";s:1:"0";}}" string(4) "b:0;" string(12) "s:5:"4.2.0";" string(1284) "a:37:{i:0;s:22:"uc_default_payment_msg";i:1;s:29:"uc_paypal_wps_checkout_button";i:2;s:23:"uc_cart_breadcrumb_text";i:3;s:17:"uc_cart_help_text";i:4;s:27:"uc_cart_new_account_details";i:5;s:24:"uc_checkout_instructions";i:6;s:31:"uc_checkout_review_instructions";i:7;s:25:"uc_continue_shopping_text";i:8;s:24:"uc_minimum_subtotal_text";i:9;s:24:"uc_msg_continue_shopping";i:10;s:26:"uc_msg_order_existing_user";i:11;s:22:"uc_msg_order_logged_in";i:12;s:21:"uc_msg_order_new_user";i:13;s:19:"uc_msg_order_submit";i:14;s:27:"uc_product_add_to_cart_text";i:15;s:26:"uc_teaser_add_to_cart_text";i:16;s:13:"uc_store_name";i:17;s:19:"uc_field_first_name";i:18;s:18:"uc_field_last_name";i:19;s:14:"uc_field_email";i:20;s:14:"uc_field_phone";i:21;s:16:"uc_field_company";i:22;s:16:"uc_field_address";i:23;s:15:"uc_field_street";i:24;s:16:"uc_field_street1";i:25;s:16:"uc_field_street2";i:26;s:13:"uc_field_city";i:27;s:13:"uc_field_zone";i:28;s:20:"uc_field_postal_code";i:29;s:16:"uc_field_country";i:30;s:33:"itweak_login_register_button_name";i:31;s:26:"itweak_login_register_name";i:32;s:32:"itweak_login_recover_button_name";i:33;s:25:"itweak_login_recover_name";i:34;s:30:"itweak_login_login_button_name";i:35;s:27:"itweak_login_username_label";i:36;s:27:"itweak_login_password_label";}" string(7) "s:0:"";" string(7) "s:0:"";" string(4) "i:0;" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(4) "i:0;" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(28) "s:20:"01/08/23 10:59:42 pm";" string(9) "s:2:"25";" string(40) "s:32:"Clear event tracking information";" string(6) "a:0:{}" string(4) "i:0;" string(158) "s:149:"The IP address %ip is banned at %site, and will not be able to access any of its content from now on. Please contact the site administrator.";" string(92) "s:84:"This host is not allowed to log in to %site. Please contact your site administrator.";" string(9) "s:2:"25";" string(9) "s:2:"25";" string(4) "i:0;" string(4) "i:1;" string(213) "s:204:"The configured threshold of %activity_threshold logins has been reached with a tota of %tracking_current_count invalid login attempts. You should review your log information about login attempts at %site.";" string(83) "s:75:"Security information: Unexpected login activity has been detected at %site.";" string(4) "i:1;" string(162) "s:153:"You have used %user_current_count out of %user_block_attempts login attempts. After all %user_block_attempts have been used, you will be unable to login.";" string(4) "b:0;" string(8) "s:1:"1";" string(82) "s:74:"The user %username has been blocked due to failed login attempts.";" string(4) "i:0;" string(153) "s:144:"The user %username (%edit_uri) has been blocked at %site due to the amount of failed login attempts. Please check the logs for more information.";" string(61) "s:53:"Security action: The user %username has been blocked.";" string(9) "s:2:"25";" string(40) "s:32:"List of all the functions in CRM";" string(8) "s:1:"0";" string(4) "i:1;" string(4) "i:0;" string(8) "s:1:"1";" string(22) "s:14:"menu-testcrm:0";" string(4) "i:0;" string(4) "i:0;" string(14) "a:1:{i:0;i:1;}" string(4) "i:0;" string(8) "s:1:"0";" string(4) "i:0;" string(4) "i:0;" string(4) "i:0;" string(15) "s:8:"comments";" string(15) "s:8:"comments";" string(15) "s:8:"comments";" string(15) "s:8:"comments";" string(15) "s:8:"comments";" string(15) "s:8:"comments";" string(15) "s:8:"comments";" string(20) "s:12:"Interactions";" string(15) "s:8:"comments";" string(15) "s:8:"comments";" string(4) "i:0;" string(4) "i:0;" string(8) "s:1:"2";" string(4) "i:0;" string(4) "i:0;" string(8) "s:1:"2";" string(4) "i:0;" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(7) "s:0:"";" string(19) "s:11:"interaction";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(19) "s:11:"interaction";" string(7) "s:0:"";" string(7) "s:0:"";" string(20) "s:12:"nodecomments";" string(20) "s:12:"nodecomments";" string(20) "s:12:"nodecomments";" string(20) "s:12:"nodecomments";" string(20) "s:12:"nodecomments";" string(20) "s:12:"nodecomments";" string(20) "s:12:"nodecomments";" string(20) "s:12:"nodecomments";" string(20) "s:12:"nodecomments";" string(20) "s:12:"nodecomments";" string(4) "i:0;" string(23) "a:1:{i:0;s:6:"status";}" string(41) "a:2:{i:0;s:6:"status";i:1;s:7:"promote";}" string(41) "a:2:{i:0;s:6:"status";i:1;s:7:"promote";}" string(41) "a:2:{i:0;s:6:"status";i:1;s:7:"promote";}" string(41) "a:2:{i:0;s:6:"status";i:1;s:7:"promote";}" string(41) "a:2:{i:0;s:6:"status";i:1;s:7:"promote";}" string(41) "a:2:{i:0;s:6:"status";i:1;s:7:"promote";}" string(42) "a:2:{i:0;s:6:"status";i:1;s:8:"revision";}" string(41) "a:2:{i:0;s:6:"status";i:1;s:7:"promote";}" string(8) "s:1:"0";" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(6) "a:0:{}" string(6) "a:0:{}" string(6) "a:0:{}" string(6) "a:0:{}" string(6) "a:0:{}" string(6) "a:0:{}" string(6) "a:0:{}" string(6) "a:0:{}" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"3";" string(8) "s:1:"3";" string(8) "s:1:"3";" string(8) "s:1:"3";" string(8) "s:1:"3";" string(8) "s:1:"3";" string(8) "s:1:"3";" string(8) "s:1:"3";" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(7) "s:0:"";" string(4) "i:0;" string(7) "s:0:"";" string(4) "i:0;" string(7) "s:0:"";" string(4) "i:1;" string(7) "s:0:"";" string(4) "i:0;" string(7) "s:0:"";" string(4) "i:1;" string(7) "s:0:"";" string(4) "i:1;" string(7) "s:0:"";" string(4) "i:1;" string(7) "s:0:"";" string(4) "i:1;" string(2) "N;" string(11) "s:4:"feed";" string(4) "b:0;" string(27) "s:19:"Rebuild permissions";" string(83) "s:75:"Hi , Please use the following link and submit feedback. [feedback-url]";" string(28) "s:20:"Request for feedback";" string(5) "i:36;" string(4) "i:0;" string(20) "s:12:"nodecomments";" string(8) "s:1:"0";" string(23) "a:1:{i:0;s:6:"enable";}" string(6) "a:0:{}" string(11) "s:4:"type";" string(11) "s:4:"each";" string(3262) "a:20:{s:11:"toggle_logo";i:1;s:11:"toggle_name";i:0;s:13:"toggle_slogan";i:0;s:14:"toggle_mission";i:1;s:24:"toggle_node_user_picture";i:0;s:27:"toggle_comment_user_picture";i:0;s:13:"toggle_search";i:0;s:14:"toggle_favicon";i:1;s:20:"toggle_primary_links";i:0;s:22:"toggle_secondary_links";i:0;s:12:"default_logo";i:0;s:9:"logo_path";s:35:"sites/default/files/strand_logo.jpg";s:11:"logo_upload";s:0:"";s:15:"default_favicon";i:1;s:12:"favicon_path";s:0:"";s:14:"favicon_upload";s:0:"";s:6:"scheme";s:39:"#0072b9,#027ac6,#2385c2,#5ab5ee,#494949";s:7:"palette";a:5:{s:4:"base";s:7:"#0072b9";s:4:"link";s:7:"#027ac6";s:3:"top";s:7:"#2385c2";s:6:"bottom";s:7:"#5ab5ee";s:4:"text";s:7:"#494949";}s:5:"theme";s:7:"garland";s:4:"info";a:10:{s:7:"schemes";a:16:{s:39:"#0072b9,#027ac6,#2385c2,#5ab5ee,#494949";s:21:"Blue Lagoon (Default)";s:39:"#464849,#2f416f,#2a2b2d,#5d6779,#494949";s:3:"Ash";s:39:"#55c0e2,#000000,#085360,#007e94,#696969";s:10:"Aquamarine";s:39:"#d5b048,#6c420e,#331900,#971702,#494949";s:17:"Belgian Chocolate";s:39:"#3f3f3f,#336699,#6598cb,#6598cb,#000000";s:10:"Bluemarine";s:39:"#d0cb9a,#917803,#efde01,#e6fb2d,#494949";s:12:"Citrus Blast";s:39:"#0f005c,#434f8c,#4d91ff,#1a1575,#000000";s:8:"Cold Day";s:39:"#c9c497,#0c7a00,#03961e,#7be000,#494949";s:9:"Greenbeam";s:39:"#ffe23d,#a9290a,#fc6d1d,#a30f42,#494949";s:11:"Mediterrano";s:39:"#788597,#3f728d,#a9adbc,#d4d4d4,#707070";s:7:"Mercury";s:39:"#5b5fa9,#5b5faa,#0a2352,#9fa8d5,#494949";s:9:"Nocturnal";s:39:"#7db323,#6a9915,#b5d52a,#7db323,#191a19";s:6:"Olivia";s:39:"#12020b,#1b1a13,#f391c6,#f41063,#898080";s:12:"Pink Plastic";s:39:"#b7a0ba,#c70000,#a1443a,#f21107,#515d52";s:12:"Shiny Tomato";s:39:"#18583d,#1b5f42,#34775a,#52bf90,#2d2d2d";s:8:"Teal Top";s:0:"";s:6:"Custom";}s:4:"copy";a:4:{i:0;s:25:"images/menu-collapsed.gif";i:1;s:29:"images/menu-collapsed-rtl.gif";i:2;s:24:"images/menu-expanded.gif";i:3;s:20:"images/menu-leaf.gif";}s:3:"css";a:1:{i:0;s:9:"style.css";}s:8:"gradient";a:4:{i:0;i:0;i:1;i:37;i:2;i:760;i:3;i:121;}s:4:"fill";a:2:{s:4:"base";a:4:{i:0;i:0;i:1;i:0;i:2;i:760;i:3;i:568;}s:4:"link";a:4:{i:0;i:107;i:1;i:533;i:2;i:41;i:3;i:23;}}s:6:"slices";a:13:{s:15:"images/body.png";a:4:{i:0;i:0;i:1;i:37;i:2;i:1;i:3;i:280;}s:17:"images/bg-bar.png";a:4:{i:0;i:202;i:1;i:530;i:2;i:76;i:3;i:14;}s:23:"images/bg-bar-white.png";a:4:{i:0;i:202;i:1;i:506;i:2;i:76;i:3;i:14;}s:17:"images/bg-tab.png";a:4:{i:0;i:107;i:1;i:533;i:2;i:41;i:3;i:23;}s:24:"images/bg-navigation.png";a:4:{i:0;i:0;i:1;i:0;i:2;i:7;i:3;i:37;}s:26:"images/bg-content-left.png";a:4:{i:0;i:40;i:1;i:117;i:2;i:50;i:3;i:352;}s:27:"images/bg-content-right.png";a:4:{i:0;i:510;i:1;i:117;i:2;i:50;i:3;i:352;}s:21:"images/bg-content.png";a:4:{i:0;i:299;i:1;i:117;i:2;i:7;i:3;i:200;}s:29:"images/bg-navigation-item.png";a:4:{i:0;i:32;i:1;i:37;i:2;i:17;i:3;i:12;}s:35:"images/bg-navigation-item-hover.png";a:4:{i:0;i:54;i:1;i:37;i:2;i:17;i:3;i:12;}s:25:"images/gradient-inner.png";a:4:{i:0;i:646;i:1;i:307;i:2;i:112;i:3;i:42;}s:8:"logo.png";a:4:{i:0;i:622;i:1;i:51;i:2;i:64;i:3;i:73;}s:14:"screenshot.png";a:4:{i:0;i:0;i:1;i:37;i:2;i:400;i:3;i:240;}}s:12:"blend_target";s:7:"#ffffff";s:13:"preview_image";s:17:"color/preview.png";s:11:"preview_css";s:17:"color/preview.css";s:10:"base_image";s:14:"color/base.png";}}" string(175195) "a:14656:{i:310;i:1;i:313;i:0;i:314;i:0;i:315;i:0;i:312;i:0;i:349;i:0;i:386;i:0;i:428;i:0;i:424;i:0;i:426;i:0;i:427;i:0;i:251;i:0;i:432;i:0;i:433;i:0;i:429;i:1;i:348;i:1;i:51;i:0;i:389;b:0;i:442;i:1;i:73;i:0;i:448;i:0;i:449;i:0;i:450;i:0;i:451;i:0;i:304;i:0;i:300;i:0;i:302;i:0;i:319;i:0;i:452;i:1;i:58;i:0;i:454;i:1;i:248;i:0;i:298;i:0;i:464;b:0;i:323;i:0;i:324;i:0;i:216;i:1;i:476;b:0;i:247;i:0;i:75;i:0;i:171;i:0;i:333;i:0;i:297;i:0;i:299;i:0;i:477;b:0;i:480;i:0;i:482;i:1;i:483;i:0;i:491;i:1;i:495;i:0;i:487;i:1;i:506;i:0;i:507;i:0;i:508;i:0;i:509;i:0;i:510;i:0;i:481;i:0;i:511;i:0;i:512;i:0;i:301;i:0;i:13;i:0;i:515;i:1;i:517;i:1;i:479;i:0;i:526;i:1;i:522;i:0;i:523;b:0;i:274;i:0;i:629;i:1;i:633;i:1;i:639;i:1;i:641;i:1;i:646;i:0;i:654;i:1;i:655;i:1;i:657;i:1;i:658;b:0;i:659;i:1;i:662;i:1;i:675;i:0;i:687;i:0;i:252;i:0;i:234;i:0;i:703;i:1;i:236;i:0;i:237;i:0;i:711;i:1;i:648;i:0;i:713;i:1;i:715;i:1;i:729;i:1;i:730;i:1;i:731;i:1;i:745;i:1;i:746;b:0;i:754;i:1;i:756;i:1;i:757;i:1;i:758;i:1;i:759;i:1;i:760;i:1;i:761;i:1;i:762;i:1;i:763;i:1;i:767;i:1;i:772;i:1;i:773;i:1;i:777;i:1;i:778;i:1;i:670;i:1;i:807;i:1;i:788;i:1;i:779;b:1;i:733;i:1;i:817;i:1;i:823;b:1;i:826;b:1;i:832;i:1;i:836;i:0;i:853;i:0;i:822;i:1;i:819;i:1;i:851;i:1;i:860;i:1;i:859;i:1;i:862;i:1;i:863;i:1;i:317;i:1;i:833;i:1;i:868;i:1;i:876;i:1;i:885;i:1;i:888;i:1;i:889;i:1;i:890;i:1;i:894;i:1;i:895;i:1;i:905;i:1;i:918;i:0;i:912;b:1;i:924;i:0;i:923;i:0;i:934;b:1;i:951;b:1;i:910;i:1;i:957;i:0;i:911;b:1;i:965;i:1;i:956;i:0;i:967;i:1;i:1007;i:0;i:1002;i:0;i:1001;i:0;i:1003;i:0;i:1006;i:0;i:294;i:0;i:1576;b:1;i:246;i:0;i:265;i:0;i:1570;b:1;i:1893;b:1;i:1859;i:1;i:1712;i:1;i:1896;i:1;i:1872;i:1;i:1910;i:1;i:1889;i:1;i:1883;i:1;i:1817;i:1;i:1820;i:1;i:1748;b:1;i:1922;b:1;i:647;i:0;i:1916;b:1;i:1930;b:1;i:1932;b:1;i:1897;i:1;i:214;i:1;i:1938;i:1;i:1939;i:0;i:60;i:0;i:1946;b:1;i:1386;i:0;i:1008;i:0;i:1775;b:1;i:1879;b:1;i:1967;b:1;i:287;i:0;i:288;i:0;i:275;i:0;i:1154;i:1;i:1153;i:0;i:1882;i:1;i:2013;i:1;i:2020;b:1;i:1731;i:1;i:1764;i:1;i:1703;i:1;i:1746;i:1;i:1715;i:1;i:1704;i:1;i:1618;i:1;i:1692;i:1;i:1659;i:1;i:10010;b:1;i:10033;b:1;i:10025;b:1;i:10019;b:1;i:10050;b:1;i:10070;b:1;i:10082;b:1;i:10103;b:1;i:10128;b:1;i:1566;i:1;i:10020;b:1;i:10018;i:1;i:286;i:0;i:10211;i:0;i:10212;i:0;i:10215;i:0;i:10217;i:0;i:10218;i:0;i:10219;i:0;i:10037;b:1;i:2019;b:1;i:10221;i:0;i:10222;i:0;i:10223;i:0;i:10224;i:0;i:10227;i:0;i:10228;i:0;i:10229;i:0;i:10233;b:1;i:10232;b:1;i:10235;b:1;i:209;i:1;i:10246;i:0;i:10236;b:1;i:10266;b:1;i:10234;i:1;i:10264;i:0;i:10273;i:1;i:1156;i:1;i:1718;i:1;i:10286;i:1;i:10424;i:1;i:1742;i:1;i:10433;i:1;i:10434;i:0;i:10435;i:0;i:10436;i:0;i:10030;i:1;i:10423;i:1;i:1888;i:1;i:10406;i:1;i:10409;i:1;i:10459;b:1;i:10466;i:1;i:10469;b:1;i:10467;b:1;i:10472;b:1;i:1472;i:1;i:10491;b:1;i:10366;i:1;i:10496;i:1;i:10497;b:1;i:10499;i:1;i:10523;i:1;i:10771;i:1;i:10187;b:1;i:10180;b:1;i:10163;b:1;i:10805;b:1;i:10806;b:1;i:10807;b:1;i:10791;b:1;i:1918;i:1;i:10829;b:1;i:10840;b:1;i:10842;b:1;i:1779;b:1;i:10860;b:1;i:10862;i:1;i:1660;i:1;i:2017;i:1;i:1700;i:1;i:1609;i:1;i:1751;i:1;i:1760;i:1;i:1752;i:1;i:1708;i:1;i:1611;i:1;i:1832;i:1;i:1721;i:1;i:1827;i:1;i:1789;i:1;i:1651;i:1;i:1591;i:1;i:1595;i:1;i:1768;i:1;i:1701;i:1;i:1766;i:1;i:1614;i:1;i:1706;i:1;i:1615;i:1;i:1727;i:1;i:1616;i:1;i:1702;i:1;i:1767;i:1;i:1728;i:1;i:1757;i:1;i:1688;i:1;i:1802;i:1;i:1777;i:1;i:1822;i:1;i:1735;i:1;i:1662;i:1;i:1667;i:1;i:1831;i:1;i:1604;i:1;i:1793;i:1;i:1593;i:1;i:1665;i:1;i:1668;i:1;i:1796;i:1;i:1776;i:1;i:1689;i:1;i:1608;i:1;i:1734;i:1;i:1680;i:1;i:1756;i:1;i:1697;i:1;i:1646;i:1;i:1654;i:1;i:1866;i:1;i:1726;i:1;i:1716;i:1;i:1803;i:1;i:1693;i:1;i:1722;i:1;i:1605;i:1;i:1781;i:1;i:1798;i:1;i:1747;i:1;i:1769;i:1;i:1694;i:1;i:1739;i:1;i:1829;i:1;i:1690;i:1;i:1687;i:1;i:1656;i:1;i:1661;i:1;i:1795;i:1;i:1648;i:1;i:1711;i:1;i:1763;i:1;i:1771;i:1;i:1707;i:1;i:1691;i:1;i:1806;i:1;i:1834;i:1;i:1773;i:1;i:1596;i:1;i:1855;i:1;i:1675;i:1;i:1784;i:1;i:1714;i:1;i:1733;i:1;i:1674;i:1;i:1799;i:1;i:10422;i:1;i:1601;i:1;i:1770;i:1;i:1597;i:1;i:1755;i:1;i:1732;i:1;i:1599;i:1;i:1862;i:1;i:1641;i:1;i:1740;i:1;i:1710;i:1;i:1669;i:1;i:1623;i:1;i:1812;i:1;i:1663;i:1;i:1813;i:1;i:1729;i:1;i:1787;i:1;i:1854;i:1;i:1686;i:1;i:1666;i:1;i:1657;i:1;i:1821;i:1;i:1637;i:1;i:1699;i:1;i:1652;i:1;i:1843;i:1;i:1737;i:1;i:1786;i:1;i:1720;i:1;i:1744;i:1;i:1857;i:1;i:1741;i:1;i:1847;i:1;i:1761;i:1;i:1807;i:1;i:1698;i:1;i:1624;i:1;i:1613;i:1;i:1643;i:1;i:1835;i:1;i:1719;i:1;i:1791;i:1;i:1871;i:1;i:1743;i:1;i:1650;i:1;i:1780;i:1;i:1808;i:1;i:1598;i:1;i:1762;i:1;i:1824;i:1;i:1851;i:1;i:1725;i:1;i:1612;i:1;i:1750;i:1;i:1792;i:1;i:1685;i:1;i:1794;i:1;i:1825;i:1;i:10869;b:1;i:10856;i:1;i:10858;i:1;i:10861;b:1;i:10666;i:1;i:10879;i:1;i:10855;i:1;i:10898;b:1;i:10901;i:0;i:10885;i:1;i:10893;i:0;i:10894;i:0;i:1647;i:1;i:10906;i:1;i:10895;i:0;i:10892;i:0;i:10896;i:1;i:1782;i:1;i:10891;i:0;i:10890;i:1;i:10878;i:1;i:10131;i:1;i:10887;i:1;i:10671;i:1;i:10889;i:0;i:10418;i:1;i:10888;i:0;i:10335;i:1;i:10594;i:1;i:10556;i:0;i:10873;i:1;i:10874;i:1;i:10797;i:1;i:10760;i:0;i:10604;i:0;i:10596;i:1;i:10608;i:0;i:10488;i:1;i:1341;i:0;i:10618;i:1;i:10614;i:1;i:10616;i:1;i:10610;i:1;i:10612;i:1;i:10527;i:1;i:10911;b:1;i:10343;i:1;i:10430;i:1;i:10345;i:1;i:10576;i:1;i:10410;i:1;i:10315;i:1;i:1505;i:0;i:1434;i:0;i:1432;i:0;i:1506;i:1;i:1512;i:1;i:1518;i:1;i:1519;i:1;i:1526;i:1;i:1552;i:1;i:10634;i:1;i:1295;i:0;i:1724;i:1;i:10387;i:1;i:10176;i:0;i:1772;i:1;i:10905;i:1;i:10920;i:1;i:10919;i:0;i:10918;i:0;i:10922;i:1;i:1206;i:1;i:10868;b:1;i:10925;b:1;i:10924;b:1;i:10897;b:1;i:1397;i:0;i:1348;i:0;i:10501;i:0;i:10514;i:0;i:10490;i:0;i:10394;i:1;i:10506;i:1;i:1785;i:1;i:1398;i:0;i:10933;i:1;i:10929;i:1;i:10910;i:0;i:10909;i:1;i:10513;i:0;i:1328;i:0;i:10507;i:0;i:10432;i:1;i:10401;i:1;i:10364;i:1;i:1362;i:0;i:1617;i:1;i:1562;i:1;i:10568;i:1;i:10944;i:1;i:10977;b:1;i:10877;i:1;i:10942;i:0;i:10945;i:1;i:10257;i:1;i:10999;b:1;i:11000;b:1;i:10998;b:1;i:11007;i:1;i:10992;i:1;i:10975;b:1;i:11016;b:1;i:10996;b:1;i:11038;b:1;i:10081;b:1;i:10159;b:1;i:10181;b:1;i:10773;b:1;i:10777;b:1;i:11048;b:1;i:11051;b:1;i:10091;i:1;i:10647;i:1;i:10515;i:1;i:11064;i:1;i:10412;b:1;i:11072;i:0;i:11076;i:1;i:10698;i:1;i:11087;i:1;i:11097;b:1;i:11078;i:1;i:11079;i:1;i:11108;b:1;i:11084;b:1;i:11116;i:1;i:11131;b:1;i:11147;b:1;i:11126;i:1;i:11149;i:1;i:11152;i:1;i:11167;b:1;i:11176;i:1;i:11166;i:0;i:11082;b:1;i:239;i:0;i:240;i:0;i:241;i:0;i:232;i:0;i:10214;i:0;i:282;i:0;i:284;i:0;i:283;i:0;i:1186;i:1;i:11205;b:1;i:11204;b:1;i:11202;b:1;i:10995;b:1;i:11214;i:1;i:11236;b:1;i:10794;i:1;i:10875;i:1;i:321;i:1;i:11115;i:0;i:11187;i:1;i:11243;i:1;i:11256;b:1;i:11264;b:1;i:1606;b:1;i:10720;b:1;i:11283;i:0;i:10213;i:0;i:11170;i:1;i:11154;i:1;i:11090;i:1;i:11045;i:1;i:11021;i:1;i:10808;i:1;i:10783;i:1;i:10039;b:1;i:10502;b:1;i:10040;b:1;i:10947;b:1;i:10935;b:1;i:11106;b:1;i:11299;b:1;i:1926;b:1;i:1928;b:1;i:1943;b:1;i:1965;b:1;i:1941;b:1;i:1971;b:1;i:1915;b:1;i:11019;b:1;i:10795;i:1;i:1905;b:1;i:1911;b:1;i:1913;b:1;i:11218;b:1;i:11094;b:1;i:11325;b:1;i:11327;b:1;i:10880;b:1;i:10853;b:1;i:10836;b:1;i:10833;b:1;i:10823;b:1;i:10963;b:1;i:10780;b:1;i:10470;b:1;i:10240;b:1;i:11273;i:1;i:11192;i:1;i:11177;i:1;i:11073;i:1;i:11005;i:1;i:11003;i:1;i:10062;i:1;i:1578;i:0;i:1903;i:1;i:1453;i:1;i:11066;b:1;i:1579;b:1;i:11063;i:1;i:11340;i:0;i:11311;b:1;i:11260;i:1;i:11200;b:1;i:11344;b:1;i:11367;b:1;i:11381;b:1;i:11341;b:1;i:11385;b:1;i:11401;b:1;i:11406;i:0;i:10994;b:1;i:253;i:0;i:11438;b:1;i:11440;i:0;i:11387;b:1;i:11422;b:1;i:11445;b:1;i:11447;b:1;i:11448;b:1;i:11443;b:1;i:11470;b:1;i:11476;b:1;i:11472;b:1;i:11473;b:1;i:11469;b:1;i:11471;b:1;i:11477;b:1;i:11478;b:1;i:11444;b:1;i:11490;b:1;i:11464;b:1;i:11520;b:1;i:11485;b:1;i:11436;b:1;i:11424;b:1;i:11539;i:0;i:11540;i:0;i:11551;b:1;i:11554;b:1;i:11503;b:1;i:11531;b:1;i:11567;b:1;i:11580;b:1;i:1197;i:1;i:1219;i:1;i:1412;i:1;i:1184;i:1;i:1223;i:1;i:1224;i:0;i:1233;i:0;i:1234;i:0;i:1237;i:1;i:1240;i:0;i:1241;i:0;i:1243;i:1;i:1302;i:1;i:1355;i:1;i:1356;i:1;i:1367;i:0;i:1387;i:1;i:1392;i:1;i:1415;i:1;i:1275;i:1;i:1331;i:0;i:11600;i:1;i:11602;i:1;i:1326;i:1;i:11545;i:1;i:1325;i:0;i:11568;i:1;i:11587;i:1;i:11592;i:1;i:11481;i:1;i:1324;i:0;i:1318;i:0;i:1317;i:0;i:1316;i:0;i:1315;i:1;i:1309;i:0;i:1308;i:0;i:1305;i:0;i:1272;i:1;i:1270;i:0;i:1268;i:1;i:1221;i:0;i:11620;i:1;i:11591;i:1;i:11590;i:1;i:11613;i:1;i:11619;b:1;i:11623;b:1;i:11392;b:1;i:10770;i:1;i:11563;i:1;i:11594;i:1;i:11608;b:1;i:11606;b:1;i:11451;i:1;i:11496;i:1;i:1334;i:1;i:10772;i:1;i:11199;i:1;i:11347;i:1;i:11625;i:1;i:11622;i:1;i:11632;i:1;i:11536;b:1;i:11522;b:1;i:11389;b:1;i:11513;b:1;i:11096;b:1;i:11642;b:1;i:11441;b:1;i:10125;i:1;i:11631;i:1;i:11675;i:0;i:11365;i:1;i:11499;b:1;i:11493;b:1;i:11681;i:1;i:10974;i:1;i:10251;i:1;i:1990;i:1;i:1539;i:1;i:1536;i:1;i:1424;i:1;i:11405;i:1;i:1321;i:1;i:11128;i:1;i:11307;i:1;i:11238;i:1;i:11240;i:1;i:11308;i:1;i:11409;i:1;i:11011;i:1;i:11130;i:1;i:10971;i:1;i:10966;i:1;i:10865;i:1;i:10457;i:1;i:10443;i:1;i:10431;i:1;i:10284;i:1;i:10282;i:1;i:10281;i:0;i:10253;i:1;i:11002;i:1;i:10190;i:1;i:1499;i:1;i:1345;i:0;i:1314;i:1;i:10156;i:1;i:10748;i:1;i:10124;i:1;i:10121;i:0;i:11259;i:1;i:10280;i:1;i:10278;i:0;i:10456;i:1;i:10569;i:1;i:11271;i:1;i:11253;i:1;i:11198;i:1;i:11008;i:1;i:1330;i:1;i:1344;i:1;i:1340;i:0;i:1338;i:0;i:1347;i:0;i:1353;i:0;i:1352;i:0;i:1417;i:0;i:1416;i:0;i:1461;i:1;i:1478;i:1;i:1515;i:1;i:1541;i:1;i:10049;i:1;i:10133;i:0;i:10153;i:1;i:10149;i:1;i:1312;i:1;i:1236;i:1;i:1246;i:0;i:1276;i:0;i:1274;i:1;i:1231;i:0;i:1292;i:0;i:1289;i:1;i:1278;i:1;i:1307;i:0;i:10203;i:0;i:10204;i:0;i:2009;i:1;i:1550;i:0;i:10123;i:0;i:11565;i:1;i:11676;i:1;i:11419;i:1;i:11420;i:1;i:11421;i:1;i:11427;i:1;i:11668;i:1;i:11495;i:1;i:11637;i:1;i:11480;i:1;i:1878;i:1;i:11460;b:1;i:11711;b:1;i:11714;i:0;i:11704;i:1;i:11715;i:1;i:11430;i:1;i:10157;i:1;i:10651;i:1;i:11720;b:1;i:11705;i:1;i:11739;i:1;i:11738;i:1;i:11736;i:1;i:11735;i:1;i:11737;i:1;i:11734;i:1;i:11621;i:1;i:11713;i:1;i:11716;i:1;i:11725;i:1;i:11733;i:1;i:11732;i:1;i:11728;i:1;i:11710;i:1;i:11706;i:1;i:11703;i:1;i:11717;i:1;i:11747;i:1;i:11750;i:1;i:11748;i:1;i:11749;i:1;i:11756;b:1;i:11763;b:1;i:11751;i:1;i:11765;b:1;i:11754;b:1;i:11781;i:1;i:10402;i:1;i:11787;b:1;i:11792;b:1;i:11804;i:1;i:11348;b:1;i:11819;b:1;i:11830;i:1;i:11831;i:1;i:11692;b:1;i:11665;b:1;i:11663;b:1;i:11657;b:1;i:11857;b:1;i:11886;i:0;i:11867;b:1;i:11865;b:1;i:11897;b:1;i:11906;b:1;i:11932;b:1;i:11800;b:1;i:11813;b:1;i:11764;b:1;i:11959;b:1;i:11961;b:1;i:11966;b:1;i:11905;b:1;i:11698;b:1;i:12021;b:1;i:12031;i:1;i:12033;i:1;i:12032;i:0;i:11922;i:1;i:11221;i:1;i:10022;i:1;i:12074;b:1;i:11505;b:1;i:11507;b:1;i:11553;b:1;i:11557;b:1;i:11626;b:1;i:11628;b:1;i:11669;b:1;i:11729;b:1;i:11758;b:1;i:11775;b:1;i:12086;b:1;i:11817;b:1;i:11821;b:1;i:11832;b:1;i:11943;b:1;i:11947;b:1;i:12099;b:1;i:12098;b:1;i:12111;b:1;i:12115;b:1;i:12116;b:1;i:12120;i:0;i:12123;i:0;i:12126;b:1;i:12061;i:1;i:12131;i:1;i:12140;i:1;i:12141;i:1;i:12148;b:1;i:11953;b:1;i:12160;b:1;i:12166;i:1;i:12149;b:1;i:12187;i:0;i:12193;b:1;i:12190;i:1;i:12191;i:1;i:12206;i:1;i:12322;b:1;i:12343;i:0;i:10675;b:1;i:12371;b:1;i:12367;b:1;i:12402;b:1;i:12406;b:1;i:12416;b:1;i:12419;b:1;i:12423;b:1;i:12360;b:1;i:12408;b:1;i:12460;b:1;i:12463;b:1;i:12497;b:1;i:12444;b:1;i:12498;b:1;i:12522;b:1;i:12519;b:1;i:12523;b:1;i:12528;b:1;i:12565;i:1;i:12568;i:1;i:12601;b:1;i:12321;b:1;i:12627;b:1;i:12536;b:1;i:12192;b:1;i:12196;b:1;i:12442;b:1;i:12465;b:1;i:12106;b:1;i:12547;b:1;i:12674;b:1;i:12692;b:1;i:12614;b:1;i:12690;b:1;i:12713;b:1;i:290;i:0;i:12740;b:1;i:12757;i:0;i:12766;b:1;i:12775;b:1;i:12664;b:1;i:12782;i:0;i:12783;i:1;i:12562;b:1;i:12806;b:1;i:12809;b:1;i:12762;b:1;i:12796;b:1;i:12844;b:1;i:12853;b:1;i:12861;i:0;i:12866;b:1;i:12524;b:1;i:12851;b:1;i:12870;b:1;i:12876;b:1;i:12874;b:1;i:12872;b:1;i:12840;b:1;i:11838;b:1;i:11881;b:1;i:11919;b:1;i:11929;b:1;i:11933;b:1;i:12916;i:1;i:12919;i:0;i:12937;b:1;i:12891;b:1;i:12945;i:0;i:12955;i:1;i:12992;i:1;i:13003;b:1;i:12813;b:1;i:12794;b:1;i:12772;b:1;i:12672;b:1;i:12677;b:1;i:12669;b:1;i:12597;b:1;i:12616;b:1;i:12560;b:1;i:12531;b:1;i:12129;b:1;i:12399;b:1;i:11901;b:1;i:13019;b:1;i:12996;b:1;i:12967;b:1;i:12828;b:1;i:12666;b:1;i:12534;b:1;i:12971;b:1;i:13028;b:1;i:12983;b:1;i:13027;b:1;i:13032;b:1;i:13065;i:1;i:13074;i:1;i:13078;b:1;i:13101;b:1;i:13136;b:1;i:13142;b:1;i:1705;i:1;i:13158;b:1;i:13181;b:1;i:13182;b:1;i:12687;b:1;i:12659;b:1;i:12172;b:1;i:10267;i:1;i:11836;b:1;i:11849;b:1;i:11917;b:1;i:11935;b:1;i:11939;b:1;i:11988;b:1;i:12007;b:1;i:12162;b:1;i:12177;b:1;i:12366;b:1;i:12373;b:1;i:12379;b:1;i:12415;b:1;i:13231;b:1;i:12491;b:1;i:12899;b:1;i:12911;b:1;i:13237;b:1;i:12927;b:1;i:12928;b:1;i:13073;b:1;i:11509;b:1;i:12097;b:1;i:12095;b:1;i:12104;b:1;i:12147;b:1;i:12317;b:1;i:12323;b:1;i:12355;b:1;i:12364;b:1;i:12470;b:1;i:12817;b:1;i:12697;b:1;i:12760;b:1;i:12978;b:1;i:13018;b:1;i:13030;b:1;i:13051;b:1;i:13077;b:1;i:13100;b:1;i:13274;b:1;i:13284;b:1;i:13283;b:1;i:13320;i:0;i:11025;i:1;i:12656;i:1;i:13362;b:1;i:13372;i:0;i:13374;i:1;i:13375;i:1;i:13377;i:0;i:13376;i:1;i:13378;i:1;i:13373;i:0;i:13380;i:0;i:13381;i:1;i:13361;b:0;i:13388;b:1;i:13312;b:0;i:13301;b:0;i:13440;i:1;i:13359;b:0;i:12802;b:0;i:12455;b:0;i:13334;b:0;i:13479;i:1;i:13392;b:1;i:1852;i:0;i:13486;i:0;i:13558;i:1;i:13600;i:1;i:13596;b:1;i:13606;i:0;i:13597;b:1;i:308;i:0;i:63;i:0;i:13616;i:0;i:10216;i:0;i:13618;i:0;i:13619;i:0;i:13620;i:0;i:13623;i:0;i:13622;i:0;i:13621;i:0;i:13624;i:0;i:13627;i:0;i:13628;i:0;i:13595;b:1;i:13097;b:0;i:13189;b:0;i:13205;b:0;i:13304;b:0;i:13632;b:1;i:13635;b:1;i:307;i:0;i:13662;b:1;i:303;i:0;i:1952;i:0;i:10208;i:0;i:285;i:0;i:13683;i:0;i:13686;i:1;i:13665;b:1;i:13668;b:1;i:11724;b:0;i:13163;b:0;i:13241;b:0;i:13296;b:0;i:13299;b:0;i:13311;b:0;i:13545;b:1;i:13642;b:1;i:13667;b:1;i:13721;b:1;i:11394;b:0;i:12018;b:0;i:11069;b:0;i:11467;b:0;i:13729;i:1;i:1497;i:0;i:1193;i:0;i:13823;i:0;i:13796;i:1;i:13865;b:1;i:13890;i:0;i:13863;b:1;i:13831;b:1;i:13934;b:1;i:13922;b:1;i:13061;i:0;i:13817;b:1;i:13480;b:1;i:11671;b:0;i:13625;b:1;i:13629;b:1;i:13758;b:1;i:13984;b:1;i:13786;b:1;i:13819;b:1;i:13734;b:1;i:13352;b:0;i:13020;b:0;i:13000;b:0;i:12855;b:0;i:12634;b:0;i:12572;b:0;i:14023;b:1;i:14030;b:1;i:14029;b:1;i:13957;i:1;i:13956;i:1;i:13873;i:1;i:12718;b:0;i:12906;b:0;i:13340;b:0;i:14087;b:1;i:14025;b:1;i:14048;b:1;i:12936;b:0;i:13348;b:0;i:14093;b:1;i:14059;b:1;i:14129;b:1;i:14146;b:1;i:14148;b:1;i:14150;b:1;i:14152;b:1;i:14164;b:1;i:14166;b:1;i:14168;b:1;i:14171;b:1;i:14173;b:1;i:14095;b:1;i:14188;i:0;i:14189;i:0;i:14191;b:1;i:14198;b:1;i:14199;b:1;i:14202;b:1;i:14201;b:1;i:14074;b:1;i:14231;b:1;i:14233;i:0;i:14234;i:0;i:14230;b:1;i:14118;b:1;i:14046;b:1;i:14322;i:1;i:14032;b:1;i:14332;i:0;i:14225;b:1;i:14022;b:1;i:14390;b:1;i:14392;b:1;i:14394;b:1;i:14398;b:1;i:14403;b:1;i:14405;b:1;i:14408;b:1;i:14411;b:1;i:14432;b:1;i:14441;b:1;i:14442;b:1;i:14446;b:1;i:14283;b:1;i:14452;b:1;i:14218;b:1;i:14458;i:0;i:14193;i:0;i:14352;b:1;i:1201;b:0;i:14470;b:1;i:14472;b:1;i:14475;b:1;i:14488;b:1;i:14490;b:1;i:14493;b:1;i:14525;b:1;i:14430;b:1;i:14522;b:1;i:14557;b:1;i:14560;b:1;i:14547;b:1;i:14573;b:1;i:14565;b:1;i:14520;b:1;i:14356;b:1;i:14644;b:1;i:14672;b:1;i:14688;b:1;i:14690;b:1;i:14514;b:1;i:14454;b:1;i:14712;b:1;i:14787;b:1;i:13833;b:1;i:14008;b:1;i:14246;b:1;i:14303;b:1;i:14243;b:1;i:14329;b:1;i:14326;b:1;i:14297;b:1;i:14824;b:1;i:14807;b:1;i:14529;b:1;i:14796;b:1;i:14504;b:1;i:14501;b:1;i:14835;b:1;i:14862;b:1;i:14878;b:1;i:14891;b:1;i:14925;b:1;i:14930;b:1;i:14934;b:1;i:14954;b:1;i:14956;i:1;i:14927;b:1;i:14995;b:1;i:14997;i:1;i:15069;b:1;i:15053;b:1;i:15086;b:1;i:13492;i:1;i:15106;b:0;i:15141;b:1;i:15062;b:0;i:15164;b:1;i:15200;b:1;i:15111;b:1;i:15208;b:1;i:14982;b:1;i:10961;b:0;i:15240;b:1;i:14973;b:1;i:14831;b:1;i:15068;b:1;i:15188;b:1;i:15291;i:0;i:15292;i:0;i:15293;i:0;i:15294;i:0;i:15295;i:0;i:15296;i:0;i:15297;i:0;i:15298;i:0;i:15299;i:0;i:15300;i:0;i:15302;i:0;i:15314;b:1;i:15310;b:1;i:15357;b:1;i:15367;b:1;i:15361;i:1;i:15376;i:0;i:15377;i:0;i:15401;i:0;i:15402;i:0;i:15445;b:1;i:15456;b:1;i:15442;b:1;i:14040;b:1;i:15471;i:1;i:15478;i:1;i:15504;i:1;i:15394;b:0;i:15431;b:1;i:15570;b:1;i:15386;b:1;i:15594;b:1;i:15566;b:1;i:15515;b:1;i:15472;b:1;i:15603;b:1;i:15619;i:1;i:15636;b:1;i:15658;b:1;i:14510;b:0;i:15752;i:0;i:15761;b:1;i:15769;i:1;i:15645;i:1;i:15600;i:1;i:15597;i:1;i:11263;b:0;i:15854;b:1;i:15855;b:1;i:15905;i:1;i:15948;b:1;i:15966;b:1;i:15976;b:1;i:15982;b:1;i:16016;b:1;i:16018;b:1;i:16021;b:1;i:16041;b:1;i:16043;b:1;i:1176;i:0;i:16052;b:1;i:16058;i:1;i:16074;b:1;i:16079;b:1;i:10324;i:1;i:16154;i:0;i:16165;b:1;i:16153;i:0;i:16197;b:1;i:16209;i:0;i:16210;i:0;i:16219;b:1;i:16225;i:0;i:16226;i:0;i:16256;i:0;i:16255;i:0;i:16292;i:1;i:16407;b:1;i:16412;i:1;i:16155;i:0;i:16468;b:1;i:16473;b:1;i:13321;b:1;i:16207;b:0;i:16459;b:1;i:16573;b:1;i:16587;i:0;i:16211;i:0;i:16618;b:1;i:16623;b:1;i:16630;b:1;i:16754;b:1;i:16769;b:1;i:16774;b:1;i:16790;b:1;i:12663;b:0;i:16811;b:1;i:16827;b:1;i:16890;i:1;i:10318;i:0;i:16927;b:1;i:15252;b:0;i:15313;b:1;i:15569;b:1;i:16077;b:1;i:17095;b:1;i:14250;b:1;i:1250;i:0;i:16824;b:0;i:17128;i:1;i:17212;b:1;i:17241;b:0;i:17288;b:1;i:17293;b:1;i:17268;b:0;i:17394;b:1;i:17407;b:1;i:17455;b:1;i:17460;b:1;i:1629;i:1;i:17547;b:1;i:17593;b:1;i:17624;b:1;i:17645;b:1;i:17676;b:1;i:17681;b:1;i:17712;b:1;i:17722;b:1;i:17762;b:1;i:17784;b:1;i:17857;b:1;i:17912;b:1;i:17950;b:1;i:17856;b:0;i:17944;b:0;i:18011;b:1;i:18046;b:1;i:18071;b:1;i:18080;b:1;i:18082;b:1;i:18084;b:1;i:17706;b:0;i:18097;b:1;i:18096;b:1;i:18095;b:1;i:18105;b:1;i:18106;b:1;i:18114;b:1;i:18113;b:1;i:18116;b:1;i:18118;b:1;i:18111;b:1;i:18117;b:1;i:18110;b:1;i:18077;b:1;i:18124;b:1;i:18109;b:1;i:18148;b:1;i:18123;b:1;i:18159;b:1;i:18157;b:1;i:18170;b:1;i:18145;b:1;i:18158;b:1;i:18177;b:1;i:18181;b:1;i:15937;b:1;i:15627;b:1;i:18179;b:1;i:18178;b:1;i:18168;b:1;i:18147;b:1;i:18186;b:1;i:18194;b:1;i:18197;b:1;i:18193;b:1;i:18141;b:1;i:18195;b:1;i:18213;b:1;i:18208;b:1;i:18207;b:1;i:18223;b:1;i:18088;b:1;i:18086;b:1;i:18137;b:1;i:18231;b:1;i:18246;b:1;i:18247;b:1;i:18245;b:1;i:18243;b:1;i:18242;b:1;i:18265;b:1;i:18268;b:1;i:18269;b:1;i:18222;b:0;i:18277;b:1;i:18280;b:1;i:18281;b:1;i:18284;b:1;i:18283;b:1;i:18264;b:1;i:18287;b:1;i:18286;b:1;i:18255;b:0;i:18040;b:1;i:18297;b:1;i:18298;b:1;i:18301;b:1;i:18300;b:1;i:18307;b:1;i:18219;b:1;i:18314;b:1;i:18313;b:1;i:18299;b:1;i:18308;i:1;i:18316;b:1;i:18324;b:1;i:18306;b:1;i:18328;b:1;i:18334;b:1;i:18257;b:1;i:18275;b:1;i:18327;b:1;i:18332;b:1;i:18348;b:1;i:18325;b:1;i:18349;b:1;i:18350;b:1;i:18244;b:1;i:18365;b:1;i:18335;b:1;i:10057;b:0;i:18363;b:1;i:18364;b:1;i:18373;b:1;i:18374;b:1;i:18387;b:1;i:18362;b:1;i:18340;b:1;i:18339;b:1;i:18256;b:1;i:18318;b:1;i:18389;b:1;i:18143;b:1;i:18393;b:1;i:18396;b:1;i:18380;b:1;i:18402;b:1;i:18408;b:1;i:18413;b:1;i:18414;b:1;i:18435;b:1;i:18425;b:1;i:18424;b:1;i:18441;b:1;i:18444;b:1;i:18447;b:1;i:18440;b:1;i:18453;i:0;i:17150;i:0;i:18457;i:0;i:18471;b:1;i:18470;b:1;i:18477;b:1;i:18476;b:1;i:18403;b:1;i:18479;b:1;i:18481;b:1;i:18409;b:1;i:18485;b:1;i:18412;b:1;i:18499;b:1;i:18404;b:1;i:18504;b:1;i:18484;b:1;i:18510;b:1;i:18488;b:1;i:18512;b:1;i:18514;b:1;i:18507;b:1;i:18533;b:1;i:18450;b:1;i:18411;b:1;i:18542;b:1;i:18545;b:1;i:18548;b:1;i:16802;b:0;i:18558;b:1;i:18555;b:1;i:18547;b:1;i:18559;b:1;i:18567;b:1;i:18529;b:1;i:18531;b:1;i:18564;b:1;i:18602;b:1;i:18534;b:1;i:18594;b:1;i:18610;b:1;i:18603;b:1;i:18600;b:1;i:18628;b:1;i:18629;b:1;i:18604;b:1;i:18626;b:1;i:18625;b:1;i:18642;b:1;i:18509;b:1;i:18656;b:1;i:18661;b:1;i:18667;b:1;i:18657;b:1;i:18691;b:1;i:18660;b:1;i:18651;b:1;i:18728;b:1;i:18725;b:1;i:18650;b:1;i:18740;b:1;i:18739;b:1;i:18726;b:1;i:18668;b:1;i:18596;b:1;i:18652;b:1;i:18751;b:1;i:18756;b:1;i:18758;b:1;i:18755;b:1;i:18781;b:1;i:18780;b:1;i:18833;b:1;i:18836;b:1;i:18729;b:1;i:18834;b:1;i:17573;b:0;i:18847;b:1;i:18832;b:1;i:18754;b:1;i:18750;b:1;i:18851;b:1;i:18864;b:1;i:16668;b:0;i:18892;b:1;i:18902;i:0;i:18903;i:0;i:14194;i:0;i:18946;b:1;i:18968;b:1;i:18976;b:1;i:18852;i:0;i:1413;b:0;i:18990;b:1;i:19000;b:1;i:19020;b:1;i:19031;b:1;i:19034;b:1;i:19036;b:1;i:19045;b:1;i:19038;b:1;i:19062;b:1;i:19164;b:1;i:18991;b:1;i:19215;b:1;i:19217;b:1;i:19201;b:1;i:19205;b:1;i:19240;b:1;i:19210;b:1;i:19252;b:1;i:19263;b:1;i:19296;b:1;i:19309;b:1;i:19314;b:1;i:19185;b:1;i:19336;b:1;i:18948;b:1;i:19351;b:1;i:19249;b:1;i:18895;b:1;i:19364;b:1;i:19366;b:1;i:19369;i:1;i:19235;b:1;i:19233;b:1;i:19237;b:1;i:19355;b:1;i:19272;b:1;i:19390;b:1;i:19403;b:1;i:19405;b:1;i:19220;b:1;i:19416;b:1;i:19401;b:1;i:19431;b:1;i:19433;b:1;i:19420;b:1;i:19392;b:1;i:19448;b:1;i:19474;b:1;i:19478;b:1;i:19473;b:1;i:19487;b:1;i:19495;b:1;i:19483;b:1;i:19511;b:1;i:19512;b:1;i:19515;b:1;i:19531;b:1;i:19529;b:1;i:19535;b:1;i:19540;b:1;i:19545;b:1;i:19494;b:1;i:19565;b:1;i:19496;b:1;i:19486;b:1;i:19484;b:1;i:19564;b:1;i:19485;b:1;i:19492;b:1;i:15810;b:0;i:19239;b:1;i:19372;b:1;i:19477;b:1;i:19572;b:1;i:19491;b:1;i:19602;b:1;i:19409;b:1;i:19612;b:1;i:19640;b:1;i:19644;b:1;i:19587;b:1;i:19537;b:1;i:19570;b:1;i:19590;b:1;i:19592;b:1;i:19667;b:1;i:19605;b:1;i:19713;b:1;i:19722;b:1;i:19738;b:1;i:19788;b:1;i:19799;b:1;i:19801;b:1;i:19703;b:1;i:19553;b:1;i:19641;b:1;i:19781;b:1;i:19782;b:1;i:19832;i:1;i:19742;b:1;i:19451;b:1;i:19857;b:1;i:19860;b:1;i:19856;b:1;i:19744;b:1;i:19837;b:1;i:19943;b:1;i:19950;b:1;i:19947;b:1;i:20001;i:1;i:20002;i:1;i:19896;b:1;i:19676;b:1;i:19898;b:1;i:20021;i:0;i:20063;i:0;i:20080;b:1;i:20083;b:1;i:19949;b:1;i:19998;i:0;i:20122;b:1;i:20150;b:1;i:20152;b:1;i:19912;i:1;i:20180;i:1;i:20196;i:1;i:20195;i:1;i:20215;b:1;i:20124;b:1;i:20239;b:1;i:20238;b:1;i:20260;b:1;i:20233;b:1;i:20245;b:1;i:20243;b:1;i:20258;b:1;i:20319;b:1;i:20303;b:1;i:20299;b:1;i:20350;i:1;i:17952;b:1;i:19639;b:1;i:19705;b:1;i:19817;b:1;i:20088;b:1;i:17403;b:1;i:17215;b:1;i:16940;b:1;i:16703;b:1;i:17449;b:1;i:17262;b:1;i:16562;b:1;i:10747;b:0;i:1639;b:0;i:20391;b:1;i:20385;b:1;i:20343;b:1;i:20420;b:1;i:20426;b:1;i:20466;b:1;i:20329;b:1;i:20342;b:1;i:20485;b:1;i:20488;b:1;i:20494;i:1;i:20484;b:1;i:20499;b:1;i:20487;b:1;i:20511;i:1;i:20477;b:1;i:20529;b:1;i:20551;b:1;i:20554;b:1;i:20555;b:1;i:20556;b:1;i:20541;b:1;i:20562;b:1;i:20593;b:1;i:20594;b:1;i:20597;b:1;i:20596;b:1;i:20604;b:1;i:20605;b:1;i:20560;b:1;i:20558;b:1;i:20613;b:1;i:20612;b:1;i:20617;b:1;i:20620;b:1;i:20623;i:0;i:20606;b:1;i:20527;b:1;i:20500;b:1;i:20643;b:1;i:20572;b:1;i:20638;b:1;i:20688;b:1;i:20690;b:1;i:20691;b:1;i:20694;b:1;i:20701;b:1;i:20709;b:1;i:20716;b:1;i:20718;b:1;i:20721;b:1;i:20724;b:1;i:20726;b:1;i:20729;b:1;i:20731;b:1;i:20733;b:1;i:20735;b:1;i:20737;b:1;i:20742;b:1;i:20745;b:1;i:20750;b:1;i:20752;b:1;i:20754;b:1;i:20758;b:1;i:20761;b:1;i:20766;b:1;i:20711;b:1;i:20706;b:1;i:20698;b:1;i:19858;b:1;i:20776;b:1;i:20807;b:1;i:20811;b:1;i:20823;b:1;i:20637;b:0;i:20830;b:1;i:20777;b:1;i:20626;b:1;i:20833;b:1;i:20836;b:1;i:20886;b:1;i:20779;b:1;i:20787;b:1;i:20923;b:1;i:20925;b:1;i:20926;b:1;i:20945;i:1;i:20946;b:1;i:20877;b:1;i:20948;b:1;i:20971;i:0;i:20964;b:1;i:20937;b:1;i:20985;b:1;i:20922;b:1;i:20832;b:1;i:20990;b:1;i:21005;b:1;i:20857;b:1;i:20809;b:1;i:21020;b:1;i:21019;b:1;i:21013;b:1;i:21000;b:1;i:21029;b:1;i:21040;i:1;i:21047;b:1;i:21055;b:1;i:21054;b:1;i:20799;b:1;i:20071;b:1;i:20074;b:1;i:20394;b:1;i:20396;b:1;i:21065;b:1;i:20521;b:1;i:20537;b:1;i:20539;b:1;i:20793;b:1;i:20789;b:1;i:21079;b:1;i:21085;b:1;i:21091;i:1;i:21092;i:1;i:21093;i:1;i:21044;b:1;i:21100;i:1;i:21101;i:1;i:21102;i:1;i:21103;i:1;i:20993;b:1;i:21046;b:1;i:21045;b:1;i:21130;b:1;i:325;i:1;i:21166;i:1;i:21171;b:0;i:21084;i:0;i:21169;b:0;i:21229;b:1;i:20965;b:0;i:21233;i:1;i:21234;i:1;i:21242;b:1;i:21243;b:1;i:21247;i:1;i:21248;i:1;i:20797;b:0;i:20358;b:0;i:20356;b:0;i:21257;b:1;i:21253;b:1;i:21261;b:1;i:21228;b:1;i:21265;b:1;i:21271;b:1;i:21270;b:1;i:21263;b:1;i:21273;i:1;i:21264;b:1;i:21277;b:1;i:20656;b:0;i:21294;b:1;i:16484;b:0;i:14426;b:0;i:14536;b:0;i:14635;b:0;i:15448;b:0;i:16086;b:0;i:16486;b:0;i:16396;b:0;i:16706;b:0;i:17028;b:0;i:17755;b:0;i:17776;b:0;i:17781;b:0;i:17815;b:0;i:17884;b:0;i:17997;b:0;i:21300;b:1;i:19387;b:0;i:19423;b:0;i:19627;b:0;i:21295;b:1;i:21319;b:1;i:21327;b:1;i:21326;b:1;i:21259;b:1;i:21338;b:1;i:21339;b:1;i:21325;b:1;i:21343;i:1;i:21321;b:1;i:19650;b:0;i:21364;b:1;i:21367;i:1;i:21381;b:1;i:21397;b:1;i:21406;i:1;i:21419;b:1;i:21421;b:1;i:21413;b:1;i:21431;b:1;i:21434;i:1;i:21438;i:1;i:21385;b:1;i:21446;b:1;i:21353;b:1;i:21427;b:1;i:21441;b:1;i:21452;b:1;i:21462;b:1;i:21467;b:1;i:21445;b:1;i:20523;b:0;i:21479;b:1;i:21480;b:1;i:21482;b:1;i:21477;b:1;i:21501;b:1;i:21508;b:1;i:21512;b:1;i:21471;b:1;i:21524;b:1;i:21456;b:1;i:21522;b:1;i:21521;b:1;i:21534;b:1;i:21539;b:1;i:21552;b:1;i:21548;b:1;i:21536;b:1;i:21537;b:1;i:21565;b:1;i:21581;b:1;i:21580;b:1;i:21531;b:1;i:21585;b:1;i:21590;i:1;i:21591;b:1;i:21596;b:1;i:21586;b:1;i:21604;b:1;i:21607;b:1;i:21559;b:1;i:21493;b:1;i:21634;b:1;i:21637;b:1;i:21639;b:1;i:21640;b:1;i:21641;i:1;i:21633;b:1;i:21660;b:1;i:21556;b:1;i:21681;b:1;i:21684;b:1;i:21672;b:1;i:21697;b:1;i:21687;b:1;i:21703;b:1;i:21705;b:1;i:21706;b:1;i:21685;b:1;i:21683;b:1;i:21712;b:1;i:21696;b:1;i:21656;b:1;i:21718;b:1;i:21707;b:1;i:21730;b:1;i:21740;b:1;i:21725;b:1;i:21719;i:1;i:21746;b:1;i:21648;b:1;i:21688;b:1;i:21778;b:1;i:21777;b:1;i:21779;b:1;i:21714;b:1;i:21774;b:1;i:21732;i:1;i:21790;b:1;i:21761;b:1;i:21511;b:1;i:21837;b:1;i:21749;b:1;i:21840;b:1;i:21839;b:1;i:21852;i:1;i:21854;b:1;i:21686;b:1;i:21823;b:1;i:21722;i:1;i:21769;b:1;i:21866;b:1;i:21742;b:1;i:21856;b:1;i:21889;b:1;i:21896;b:1;i:21922;b:1;i:21932;b:1;i:21931;b:1;i:21930;b:1;i:21940;b:1;i:21951;b:1;i:21959;b:1;i:21960;b:1;i:21897;b:1;i:21965;b:1;i:21966;b:1;i:21971;b:1;i:21984;b:1;i:21999;b:1;i:22003;b:1;i:22014;b:1;i:21998;b:1;i:22017;b:1;i:22019;b:1;i:22031;b:1;i:22035;b:1;i:22033;b:1;i:22037;b:1;i:22038;b:1;i:22043;b:1;i:22045;b:1;i:22051;b:1;i:22055;b:1;i:22065;b:1;i:22071;b:1;i:22061;b:1;i:22075;b:1;i:22077;b:1;i:22076;i:1;i:22057;b:1;i:22079;b:1;i:22059;b:1;i:21899;b:1;i:22086;b:1;i:22093;b:1;i:22073;b:1;i:22006;b:1;i:22108;b:1;i:22090;b:1;i:22110;b:1;i:22109;b:1;i:22088;b:1;i:22047;b:1;i:22125;b:1;i:22141;i:1;i:1632;i:1;i:1634;i:1;i:1622;i:0;i:22157;b:1;i:22152;b:1;i:22159;b:1;i:22162;b:1;i:22164;b:1;i:22166;b:1;i:22176;b:1;i:22174;b:1;i:19317;b:0;i:21052;b:0;i:21892;b:1;i:22063;b:1;i:22200;b:1;i:15032;b:0;i:22202;b:1;i:22187;b:1;i:22257;i:0;i:22258;b:1;i:22265;b:1;i:12349;i:1;i:22196;b:1;i:19383;b:0;i:18868;b:0;i:15123;b:0;i:16005;b:0;i:16007;b:0;i:18870;b:0;i:19155;b:0;i:19191;b:0;i:22116;b:1;i:22297;i:1;i:22254;b:1;i:1403;i:0;i:18290;i:1;i:22299;i:1;i:22314;b:1;i:22284;i:1;i:22289;b:1;i:22328;b:1;i:22324;b:1;i:22330;b:1;i:22331;b:1;i:22344;b:1;i:22349;b:1;i:22365;i:0;i:22361;i:1;i:22380;b:1;i:22383;b:1;i:22387;b:1;i:22177;b:1;i:21423;b:1;i:22205;b:1;i:21818;b:1;i:21821;b:1;i:21825;b:1;i:21924;b:1;i:22206;b:1;i:22209;b:1;i:22261;b:1;i:22352;b:1;i:22381;b:1;i:22414;b:1;i:22374;b:1;i:22423;b:1;i:22401;i:1;i:22403;b:1;i:22402;b:1;i:22369;b:1;i:22364;i:1;i:22431;i:1;i:22430;b:1;i:22441;b:1;i:22444;b:1;i:22445;b:1;i:22443;b:1;i:22455;b:1;i:22458;i:1;i:22459;b:1;i:22139;b:1;i:22405;i:1;i:22475;b:1;i:22479;b:1;i:22434;b:1;i:22453;b:1;i:14320;i:0;i:16174;i:0;i:22515;i:1;i:22354;b:1;i:22510;i:1;i:22529;b:1;i:18622;i:1;i:18620;i:1;i:18619;i:0;i:18618;i:1;i:18617;i:0;i:18615;i:0;i:18614;i:1;i:18613;i:0;i:18612;i:0;i:18616;i:0;i:22530;b:1;i:22546;b:1;i:22545;b:1;i:22547;b:1;i:22527;b:1;i:22512;b:1;i:22548;b:1;i:20461;b:0;i:20424;b:0;i:22558;b:1;i:22560;b:1;i:20464;b:0;i:22564;b:1;i:20468;b:0;i:22571;b:1;i:22586;b:1;i:21018;b:0;i:22598;b:1;i:22602;b:1;i:22603;b:1;i:22552;b:1;i:22553;b:1;i:22551;b:1;i:22550;b:1;i:22613;b:1;i:22608;b:1;i:22543;b:1;i:22604;b:1;i:22626;i:0;i:22622;b:1;i:22638;b:1;i:22627;b:1;i:22642;b:1;i:22651;b:1;i:22659;b:1;i:22665;i:0;i:22624;b:1;i:22669;i:1;i:22451;b:1;i:22263;b:1;i:22121;b:1;i:21914;b:1;i:21842;b:1;i:21464;b:1;i:21323;b:1;i:20998;b:0;i:22708;b:1;i:22436;i:1;i:22721;b:1;i:22709;i:1;i:22621;b:1;i:22439;b:1;i:22726;b:1;i:22741;b:1;i:22744;b:1;i:22750;b:1;i:22752;b:1;i:22461;b:1;i:22765;b:1;i:22774;b:1;i:22771;b:1;i:22769;i:1;i:22763;i:1;i:22764;b:1;i:22760;i:1;i:22762;b:1;i:22781;b:1;i:22788;b:1;i:22667;b:1;i:22818;b:1;i:22820;b:1;i:22822;b:1;i:22795;b:1;i:22680;b:1;i:22828;b:1;i:22834;b:1;i:21802;b:1;i:22844;b:1;i:22852;b:1;i:22817;i:1;i:22678;b:1;i:22829;b:1;i:22864;i:1;i:22865;b:1;i:22697;b:1;i:22856;i:1;i:22881;b:1;i:22884;b:1;i:22886;b:1;i:22891;i:1;i:22899;b:1;i:22898;i:1;i:22883;b:1;i:22906;b:1;i:22915;b:1;i:22921;b:1;i:22926;b:1;i:22612;b:1;i:22875;b:1;i:22876;b:1;i:22903;b:1;i:22900;b:1;i:22943;b:1;i:22730;b:1;i:22683;b:1;i:22954;b:1;i:22953;b:1;i:22955;b:1;i:22961;b:1;i:22972;i:1;i:22975;b:1;i:22794;b:1;i:22924;b:1;i:23008;b:1;i:22977;b:1;i:23030;b:1;i:23031;b:1;i:23032;b:1;i:21255;b:1;i:21469;b:1;i:21474;b:1;i:21515;b:1;i:21782;b:1;i:21996;b:1;i:22648;b:1;i:22928;b:1;i:22772;b:1;i:21147;i:1;i:21146;i:1;i:23055;b:1;i:22559;b:1;i:23028;i:1;i:23057;b:1;i:23064;b:1;i:22690;b:1;i:22595;b:1;i:22630;b:1;i:23069;b:1;i:22823;b:1;i:23060;i:1;i:23093;b:1;i:23089;b:1;i:23094;b:1;i:23056;b:1;i:22911;b:1;i:23010;b:1;i:23113;b:1;i:23119;b:1;i:23129;b:1;i:23134;i:1;i:23109;b:1;i:23142;b:1;i:23038;b:1;i:23036;b:1;i:23133;b:1;i:23128;b:1;i:23132;b:1;i:23160;b:1;i:23156;b:1;i:23168;b:1;i:23169;b:1;i:23155;b:1;i:23177;i:1;i:23175;b:1;i:23162;i:1;i:23186;b:1;i:23188;b:1;i:23015;b:1;i:23196;b:1;i:23200;b:1;i:23202;b:1;i:23191;b:1;i:23206;b:1;i:23091;b:1;i:23212;b:1;i:23203;b:1;i:23101;b:1;i:23153;b:1;i:23209;b:1;i:23227;b:1;i:23228;b:1;i:23229;b:1;i:23226;b:1;i:23223;b:1;i:23240;b:1;i:23243;b:1;i:23249;b:1;i:23232;b:1;i:23246;b:1;i:23245;b:1;i:23261;b:1;i:23268;i:0;i:23271;b:1;i:23274;b:1;i:23255;b:1;i:23290;b:1;i:23174;b:1;i:23294;b:1;i:23301;b:1;i:23269;b:1;i:23293;b:1;i:23225;b:1;i:23190;b:1;i:23216;b:1;i:23315;b:1;i:23298;b:1;i:23319;b:1;i:23320;i:0;i:23314;b:1;i:20077;b:0;i:22386;b:1;i:22388;b:1;i:22415;b:1;i:22484;b:1;i:22486;b:1;i:22522;b:1;i:22729;b:1;i:22758;b:1;i:22767;b:1;i:23307;b:1;i:23313;b:1;i:23372;b:1;i:22749;b:1;i:23165;b:1;i:23391;b:1;i:23392;b:1;i:23398;b:1;i:23405;i:1;i:23407;b:1;i:23409;i:1;i:23412;b:1;i:23411;b:1;i:22710;b:1;i:22806;b:1;i:20941;b:0;i:20448;b:0;i:20226;b:0;i:20194;b:0;i:19955;b:0;i:19953;b:0;i:19843;b:0;i:19842;b:0;i:19562;b:0;i:19558;b:0;i:19276;b:0;i:18938;b:0;i:18826;b:0;i:18683;b:0;i:18344;b:0;i:18251;b:0;i:18211;b:0;i:18154;b:0;i:21878;b:1;i:17987;b:0;i:17932;b:0;i:17931;b:0;i:17880;b:0;i:17879;b:0;i:17873;b:0;i:17715;b:0;i:17691;b:0;i:17988;b:0;i:17566;b:0;i:23414;b:1;i:23413;b:1;i:23416;b:1;i:23417;b:1;i:23423;b:1;i:23422;i:1;i:23397;b:1;i:23418;i:1;i:23428;b:1;i:23253;b:1;i:23373;b:1;i:23431;i:1;i:23434;b:1;i:23477;b:1;i:23436;i:1;i:23480;b:1;i:23399;i:1;i:23491;b:1;i:23494;b:1;i:23501;b:1;i:23505;b:1;i:23521;b:1;i:23526;b:1;i:23527;b:1;i:23517;b:1;i:23534;b:1;i:23557;b:1;i:23559;b:1;i:23556;b:1;i:23599;b:1;i:23604;b:1;i:23615;b:1;i:23621;i:0;i:23624;i:1;i:23625;i:1;i:23626;b:1;i:23627;i:1;i:23653;i:1;i:23658;i:1;i:23659;b:1;i:23672;b:1;i:23677;b:1;i:23683;b:1;i:23685;b:1;i:23680;b:1;i:23692;b:1;i:23700;b:1;i:23708;b:1;i:23646;b:1;i:23721;b:1;i:23731;b:1;i:23730;b:0;i:23733;b:1;i:23739;b:1;i:23741;b:1;i:23686;b:1;i:22633;b:1;i:22917;b:1;i:22682;b:1;i:22922;b:1;i:23375;b:1;i:23377;b:1;i:23379;b:1;i:23381;b:1;i:23402;b:1;i:23774;b:1;i:23734;b:1;i:23779;b:1;i:23751;b:1;i:23793;b:1;i:23790;b:1;i:23792;b:1;i:23750;b:1;i:18225;b:0;i:21792;b:1;i:21506;b:1;i:21503;b:1;i:23787;b:1;i:23780;b:1;i:23839;b:1;i:23843;b:1;i:23622;b:1;i:23845;b:1;i:23846;b:1;i:23852;b:1;i:23884;b:1;i:23886;b:1;i:23847;b:1;i:23893;b:1;i:23896;b:1;i:23899;b:1;i:23901;b:1;i:23909;b:1;i:23908;b:1;i:23910;b:1;i:23895;b:1;i:23939;b:1;i:23944;i:1;i:23946;b:1;i:23951;b:1;i:23931;b:1;i:23952;b:1;i:23956;b:1;i:23961;b:1;i:23993;b:1;i:23995;b:1;i:23881;b:1;i:24016;b:1;i:24031;b:1;i:24030;b:1;i:24047;b:1;i:24051;b:1;i:23996;b:1;i:24052;b:1;i:24036;i:1;i:24054;b:1;i:24064;b:1;i:24065;b:1;i:24029;b:1;i:24015;b:1;i:24069;i:1;i:24072;b:1;i:24046;b:1;i:24071;i:1;i:10035;i:0;i:23860;b:1;i:24061;b:1;i:24083;b:1;i:24081;b:1;i:24096;b:1;i:24094;b:1;i:24109;b:1;i:24118;b:1;i:24124;b:1;i:24093;i:1;i:24132;b:1;i:24148;b:1;i:24149;b:1;i:23948;b:1;i:24165;b:1;i:24169;b:1;i:24168;i:1;i:24177;b:1;i:24174;b:1;i:24164;b:1;i:24167;b:1;i:24189;b:1;i:24197;b:1;i:15719;b:0;i:24057;b:1;i:24179;b:1;i:24228;b:1;i:24186;b:1;i:24232;b:1;i:24234;b:1;i:24233;b:1;i:24244;b:1;i:24097;b:1;i:24259;i:1;i:24225;b:1;i:24278;b:1;i:24237;b:1;i:24292;b:0;i:24296;b:1;i:24298;b:1;i:24235;b:1;i:24183;b:1;i:24275;b:1;i:24313;b:1;i:24315;b:1;i:24330;b:1;i:24334;b:1;i:24339;b:1;i:24144;b:1;i:24178;b:1;i:24355;b:1;i:24367;b:1;i:24366;b:1;i:24368;b:1;i:24370;b:1;i:24299;b:1;i:24371;b:1;i:24376;i:1;i:24375;b:1;i:24346;i:1;i:24384;b:1;i:24383;b:1;i:24391;b:1;i:24363;b:1;i:24390;b:1;i:24392;b:1;i:24401;b:1;i:24304;b:1;i:24403;b:1;i:24193;b:1;i:24191;b:1;i:24404;b:1;i:24397;b:1;i:24416;b:1;i:24344;i:1;i:24427;i:1;i:24423;b:1;i:23329;b:1;i:23484;b:1;i:24053;b:1;i:24120;b:1;i:24122;b:1;i:24211;b:1;i:24102;b:1;i:24100;b:1;i:24091;b:1;i:23932;b:1;i:23752;b:1;i:24442;i:1;i:24418;b:1;i:24446;b:1;i:23619;b:1;i:23164;b:1;i:22273;b:1;i:22066;b:1;i:24295;b:1;i:24373;b:1;i:24459;b:1;i:24410;b:1;i:24476;b:1;i:24445;b:1;i:24327;b:1;i:24482;b:1;i:24494;i:1;i:24497;i:1;i:24498;b:1;i:24467;b:1;i:24491;i:1;i:24496;i:1;i:24464;i:1;i:24452;b:1;i:24322;b:1;i:24488;b:1;i:24320;b:1;i:24533;b:1;i:24534;b:1;i:24535;b:1;i:24540;b:1;i:24543;b:1;i:24546;b:1;i:24551;b:1;i:24565;b:1;i:24560;b:1;i:24557;b:1;i:24553;b:1;i:24549;b:1;i:24569;b:1;i:24575;b:1;i:24578;b:1;i:24580;b:1;i:24586;b:1;i:24591;b:1;i:24593;b:1;i:24603;b:1;i:24602;b:1;i:24606;b:1;i:24612;i:1;i:24616;b:1;i:24620;b:1;i:24625;b:1;i:24607;b:1;i:24610;b:1;i:24608;b:1;i:24634;b:1;i:24597;b:1;i:24461;b:1;i:24659;b:1;i:24664;b:1;i:24658;b:1;i:24655;b:1;i:24663;b:1;i:24672;b:1;i:24673;b:1;i:24676;b:1;i:24671;b:1;i:24680;i:0;i:24681;b:1;i:24525;b:1;i:24694;b:1;i:24695;b:1;i:24693;b:1;i:24711;b:1;i:24717;b:1;i:24716;b:1;i:24726;b:1;i:24729;b:1;i:24730;b:1;i:24714;b:1;i:24718;b:1;i:24738;b:1;i:24624;b:1;i:24745;b:1;i:24749;b:1;i:24746;b:1;i:24752;b:1;i:24754;b:1;i:24755;b:1;i:24757;b:1;i:24759;b:1;i:24720;b:1;i:24767;b:1;i:24699;b:1;i:24776;b:1;i:24778;b:1;i:24743;b:1;i:24799;b:1;i:24808;b:1;i:24807;i:1;i:24812;b:1;i:24791;i:1;i:24814;i:1;i:24816;b:1;i:24821;b:1;i:24810;b:1;i:24409;b:1;i:24820;b:1;i:24822;b:1;i:24740;i:1;i:24825;b:1;i:24758;i:1;i:24827;b:1;i:24832;b:1;i:24841;b:1;i:24840;i:1;i:24647;b:1;i:24697;b:1;i:24849;b:1;i:24851;i:0;i:24852;b:1;i:24867;i:0;i:24873;b:1;i:24877;b:1;i:24875;b:1;i:24878;b:1;i:24859;i:1;i:24891;b:1;i:24890;b:1;i:24893;i:0;i:24905;b:1;i:24903;b:1;i:24914;b:1;i:24915;b:1;i:24450;b:1;i:24946;b:1;i:24947;b:1;i:24954;i:1;i:24957;b:1;i:24961;i:1;i:24965;b:1;i:24977;i:1;i:24982;b:1;i:24988;b:1;i:24952;b:1;i:24997;b:1;i:24939;b:1;i:24881;b:1;i:24920;b:1;i:24887;i:1;i:24775;b:1;i:24398;i:1;i:25043;i:1;i:25048;b:1;i:25007;b:1;i:24959;b:1;i:25006;b:1;i:25055;b:1;i:25056;b:1;i:25047;b:1;i:25057;i:1;i:25067;i:1;i:25070;b:1;i:24975;b:1;i:25063;i:1;i:25059;i:1;i:25003;b:1;i:25046;b:1;i:25092;b:1;i:25093;b:1;i:25104;b:1;i:25106;b:1;i:25114;b:1;i:25112;b:1;i:25100;i:1;i:25120;b:1;i:25050;b:1;i:25123;b:1;i:24468;b:1;i:25136;b:1;i:25133;i:1;i:25139;b:1;i:25147;i:1;i:24527;b:1;i:25164;b:1;i:25166;b:1;i:25169;b:1;i:25168;i:1;i:25173;i:1;i:25172;i:1;i:25178;b:1;i:25182;b:1;i:25184;b:1;i:25177;b:1;i:25192;b:1;i:25208;b:1;i:25210;b:1;i:25156;b:1;i:25216;b:1;i:25209;b:1;i:24785;b:1;i:25234;b:1;i:25235;i:1;i:25236;b:1;i:25189;b:1;i:25237;b:1;i:25240;i:1;i:25241;i:1;i:25253;b:1;i:25258;i:1;i:25219;b:1;i:25207;b:1;i:25214;b:1;i:25212;b:1;i:25264;b:1;i:23039;b:1;i:23150;b:1;i:25274;b:1;i:25286;b:1;i:25291;b:1;i:25299;i:1;i:25181;i:1;i:24884;i:1;i:25358;b:1;i:25377;i:1;i:25380;b:1;i:25370;b:1;i:25394;i:1;i:25382;b:1;i:25384;b:1;i:25401;b:1;i:25405;b:1;i:25102;b:1;i:25391;b:1;i:25407;i:1;i:25409;i:1;i:25412;i:1;i:24594;b:1;i:24599;b:1;i:20768;b:0;i:24973;b:1;i:25435;b:1;i:15753;i:0;i:25453;b:1;i:25452;b:1;i:24762;b:1;i:24760;b:1;i:25270;b:1;i:25464;b:1;i:25465;b:1;i:25439;b:1;i:25473;b:1;i:25476;b:1;i:25487;b:1;i:25227;b:1;i:25441;b:1;i:25474;b:1;i:25472;b:1;i:25504;b:1;i:25486;b:1;i:25392;b:1;i:25527;b:1;i:25502;b:1;i:25542;b:1;i:25535;b:1;i:25545;b:1;i:25546;b:1;i:25544;b:1;i:25568;b:1;i:25572;b:0;i:25579;b:1;i:25511;b:1;i:25108;b:1;i:25596;b:1;i:25573;b:1;i:25597;b:1;i:25575;b:1;i:25569;b:1;i:25604;b:1;i:25608;b:1;i:25615;b:1;i:25625;b:1;i:25491;b:1;i:25628;b:1;i:25490;b:1;i:25631;b:1;i:25629;b:1;i:25632;b:1;i:25633;b:1;i:25555;b:1;i:25645;b:1;i:25627;b:1;i:25648;b:1;i:25649;b:1;i:25654;b:1;i:25326;b:1;i:25666;b:1;i:25085;b:1;i:25668;b:1;i:25671;b:1;i:25673;b:1;i:25643;b:1;i:25677;i:1;i:25689;b:1;i:25687;b:1;i:25697;b:1;i:25698;b:1;i:25655;b:1;i:25709;b:1;i:25719;b:1;i:25723;i:1;i:25722;b:1;i:25585;b:1;i:25592;b:1;i:25350;b:1;i:25345;b:1;i:25288;b:1;i:25267;b:1;i:25129;b:1;i:25065;b:1;i:25010;b:1;i:25755;b:1;i:25757;b:1;i:25759;b:1;i:25635;b:1;i:25729;b:1;i:25619;b:1;i:25618;b:1;i:25770;b:1;i:25768;b:1;i:25784;b:1;i:25776;b:1;i:25788;b:1;i:25797;b:1;i:25801;b:1;i:25808;b:1;i:25819;i:1;i:25798;b:1;i:24804;i:1;i:25835;b:1;i:25843;b:1;i:25820;b:1;i:25846;b:1;i:25853;b:1;i:25851;i:1;i:25855;b:1;i:25807;b:1;i:25860;b:1;i:24668;b:1;i:24529;b:1;i:25825;i:1;i:25879;b:1;i:23561;b:1;i:25600;b:1;i:25901;b:1;i:25906;b:1;i:25905;b:1;i:25918;b:1;i:25919;b:1;i:25936;b:1;i:25946;b:1;i:25925;b:1;i:25956;b:1;i:25959;b:1;i:25800;b:1;i:25964;b:1;i:25538;b:1;i:25969;i:1;i:25974;i:1;i:25976;i:1;i:25977;i:1;i:25983;b:1;i:25998;b:1;i:26001;b:1;i:25899;b:1;i:26008;b:0;i:26009;i:1;i:26010;b:1;i:26023;b:1;i:25893;b:1;i:25932;b:1;i:25989;b:1;i:26005;b:1;i:26032;b:0;i:25866;b:1;i:26031;i:1;i:26035;b:1;i:26070;b:1;i:26065;b:1;i:25995;b:1;i:26081;b:1;i:26086;i:1;i:25903;b:1;i:26091;b:1;i:26090;b:1;i:26027;b:1;i:26017;b:1;i:26058;b:1;i:26045;b:1;i:26099;b:1;i:26042;b:1;i:26126;b:1;i:26037;b:1;i:26119;b:1;i:26123;b:1;i:26140;b:1;i:26166;b:1;i:26170;b:1;i:26174;b:1;i:26125;b:1;i:26187;i:1;i:26136;b:1;i:26049;b:1;i:25870;b:1;i:25868;b:1;i:25823;b:1;i:26196;b:1;i:26197;b:1;i:26203;b:1;i:26205;b:1;i:26115;b:1;i:26207;b:1;i:26186;b:1;i:26210;i:1;i:26211;i:1;i:26212;b:1;i:26216;i:1;i:26226;b:1;i:26227;b:1;i:26240;b:1;i:25896;b:1;i:26181;b:1;i:26255;b:1;i:26252;i:1;i:26184;b:1;i:26239;i:1;i:26208;b:1;i:26297;b:1;i:26287;b:1;i:26274;b:1;i:26267;b:1;i:26322;b:1;i:26326;b:1;i:25704;b:1;i:25706;b:1;i:26337;b:1;i:26341;b:1;i:26342;b:1;i:26317;b:1;i:26348;b:1;i:26175;b:1;i:26361;b:1;i:26375;b:1;i:26374;b:0;i:26397;b:1;i:26395;b:1;i:26398;b:1;i:26400;b:1;i:26330;i:1;i:25515;b:1;i:25690;b:1;i:25692;b:1;i:25751;b:1;i:25780;b:1;i:25938;b:1;i:26109;b:1;i:26414;b:1;i:26422;i:1;i:26381;i:1;i:26425;i:1;i:26416;b:1;i:26415;b:1;i:26411;b:1;i:26410;b:1;i:26413;b:1;i:26449;b:1;i:26445;b:1;i:26454;b:1;i:26448;b:1;i:26440;b:1;i:26439;b:1;i:26467;b:1;i:26482;b:1;i:26479;b:1;i:26444;b:1;i:26493;b:1;i:26497;b:1;i:26494;b:1;i:26179;b:1;i:26498;b:1;i:26502;b:1;i:26486;b:1;i:26453;b:1;i:26358;b:1;i:26528;b:1;i:26529;b:1;i:26509;b:1;i:26542;b:1;i:26429;i:1;i:26457;b:1;i:26565;b:1;i:26560;b:1;i:26574;b:1;i:26517;b:1;i:26355;b:1;i:26488;b:1;i:26462;b:1;i:26575;b:1;i:26595;b:1;i:26593;b:1;i:26596;b:1;i:26600;i:1;i:26604;b:1;i:26612;b:1;i:26609;b:1;i:26611;b:1;i:26607;b:1;i:26614;b:1;i:26547;b:1;i:26618;b:1;i:26619;b:1;i:26620;b:1;i:26623;b:1;i:26622;i:1;i:26624;b:1;i:26554;b:1;i:26587;b:1;i:25454;b:1;i:25553;b:1;i:26635;b:1;i:26640;b:1;i:26581;b:1;i:26480;b:1;i:26570;b:1;i:26650;b:1;i:26661;b:1;i:26663;b:1;i:26658;b:1;i:26677;b:1;i:26654;b:1;i:26681;b:1;i:26701;b:1;i:26685;b:1;i:26659;b:1;i:26723;b:1;i:26679;b:1;i:26731;b:1;i:26732;b:1;i:25229;b:1;i:25557;b:1;i:25637;b:1;i:25789;i:1;i:25791;b:1;i:26583;b:1;i:26688;i:1;i:26751;i:1;i:25949;b:1;i:26760;b:1;i:26666;i:1;i:26753;b:1;i:26771;b:1;i:26773;b:1;i:26776;b:1;i:26775;b:1;i:26763;b:1;i:26541;b:1;i:26782;i:1;i:26797;i:1;i:26700;b:1;i:26802;b:1;i:26801;b:1;i:26543;b:1;i:26811;b:1;i:26730;b:1;i:26823;b:1;i:26825;b:1;i:26820;b:1;i:26834;b:1;i:26833;b:1;i:26812;b:1;i:26804;b:1;i:26851;i:1;i:26778;i:1;i:26783;b:1;i:26788;b:1;i:26824;b:1;i:26868;b:1;i:26867;b:0;i:26872;b:1;i:26873;b:1;i:26869;b:1;i:26871;b:1;i:26870;b:1;i:26842;b:1;i:26847;b:1;i:26849;b:1;i:26890;b:1;i:26878;b:1;i:26901;b:1;i:26916;b:1;i:26912;b:1;i:26931;b:1;i:26933;b:1;i:26930;b:1;i:26882;i:1;i:26902;b:1;i:26924;b:0;i:26923;b:0;i:26919;b:0;i:26921;b:0;i:26945;b:1;i:26959;b:1;i:26908;b:1;i:26975;b:1;i:26974;b:1;i:26980;b:1;i:26952;b:1;i:26988;b:1;i:26960;b:1;i:26951;b:1;i:26996;b:1;i:26826;b:1;i:26942;b:1;i:26915;b:1;i:26968;b:1;i:27007;b:1;i:27014;b:1;i:26997;b:1;i:27024;b:1;i:27027;b:1;i:26676;b:1;i:27043;b:1;i:26987;i:1;i:27020;b:1;i:27037;b:1;i:27049;i:1;i:26947;b:1;i:26954;b:1;i:26956;b:1;i:26955;b:1;i:27087;b:1;i:27092;b:1;i:27097;b:1;i:27098;b:1;i:27047;b:1;i:27103;b:1;i:19815;b:0;i:27111;b:1;i:27112;b:1;i:27115;b:1;i:27116;b:1;i:27124;b:1;i:27096;b:1;i:27131;b:1;i:26925;b:1;i:26998;b:1;i:27057;b:1;i:27075;b:0;i:27146;b:1;i:27185;b:1;i:27191;b:1;i:25498;b:1;i:27197;b:1;i:27134;b:1;i:27226;b:1;i:27228;i:1;i:27153;b:1;i:27237;i:1;i:27238;b:1;i:27166;b:1;i:27213;b:1;i:27276;b:1;i:27280;b:1;i:27283;b:1;i:27286;b:1;i:27315;b:1;i:27328;b:1;i:27330;b:1;i:27338;b:1;i:27287;b:1;i:27348;b:1;i:27003;b:1;i:27322;b:1;i:27354;b:1;i:27356;b:1;i:27367;b:1;i:27365;i:1;i:26887;b:1;i:27379;b:1;i:27387;b:1;i:27399;b:1;i:27327;b:1;i:27403;b:1;i:27193;b:1;i:27426;i:1;i:27452;i:1;i:27455;b:1;i:27372;b:1;i:27419;i:1;i:27473;b:1;i:27472;i:1;i:27476;b:1;i:27454;b:1;i:27480;b:1;i:27487;b:1;i:27486;b:1;i:27453;b:1;i:27496;b:1;i:27501;b:1;i:21678;b:1;i:19353;i:0;i:19425;i:0;i:16994;b:0;i:27506;b:1;i:27503;b:1;i:27513;b:1;i:17311;b:0;i:16874;b:0;i:16863;b:0;i:16861;b:0;i:16840;b:0;i:16760;b:0;i:16723;b:0;i:16738;b:0;i:16674;b:0;i:16677;b:0;i:16672;b:0;i:16680;b:0;i:16604;b:0;i:16579;b:0;i:16545;b:0;i:16541;b:0;i:16577;b:0;i:16426;b:0;i:16575;b:0;i:27517;b:1;i:27521;b:1;i:16408;b:0;i:16398;b:0;i:16279;b:0;i:15980;b:0;i:15894;b:0;i:15862;b:0;i:15901;b:0;i:15860;b:0;i:15832;b:0;i:15830;b:0;i:15845;b:0;i:15840;b:0;i:15834;b:0;i:15847;b:0;i:15734;b:0;i:15732;b:0;i:15713;b:0;i:15721;b:0;i:15716;b:0;i:15756;b:0;i:15723;b:0;i:15727;b:0;i:15678;b:0;i:15655;b:0;i:15634;b:0;i:15625;b:0;i:15642;b:0;i:15640;b:0;i:15630;b:0;i:15632;b:0;i:15554;b:0;i:15549;b:0;i:15552;b:0;i:15546;b:0;i:14402;b:0;i:15003;b:0;i:27271;b:1;i:27298;b:1;i:27505;b:1;i:27345;b:1;i:27492;b:1;i:27544;b:1;i:27548;b:1;i:27450;b:1;i:27554;b:1;i:27362;b:1;i:27448;b:1;i:27518;b:1;i:27577;b:1;i:27580;b:1;i:27578;b:1;i:27581;i:1;i:27595;b:1;i:27596;b:1;i:27105;b:1;i:27562;b:1;i:14330;b:0;i:14369;b:0;i:14372;b:0;i:14374;b:0;i:14376;b:0;i:14534;b:0;i:14579;b:0;i:14538;b:0;i:14553;b:0;i:14555;b:0;i:14599;b:0;i:14669;b:0;i:14677;b:0;i:14699;b:0;i:14710;b:0;i:14717;b:0;i:14857;b:0;i:14895;b:0;i:14978;b:0;i:14906;b:0;i:14919;b:0;i:14912;b:0;i:15028;b:0;i:14915;b:0;i:14917;b:0;i:15024;b:0;i:15026;b:0;i:14975;b:0;i:14970;b:0;i:15022;b:0;i:15020;b:0;i:15036;b:0;i:15039;b:0;i:15011;b:0;i:15014;b:0;i:15016;b:0;i:15017;b:0;i:15030;b:0;i:15043;b:0;i:15045;b:0;i:15047;b:0;i:15050;b:0;i:15080;b:0;i:15084;b:0;i:15079;b:0;i:15081;b:0;i:15195;b:0;i:15171;b:0;i:15181;b:0;i:27609;b:1;i:27587;b:1;i:27623;b:1;i:15143;b:0;i:15145;b:0;i:15197;b:0;i:15193;b:0;i:15205;b:0;i:15166;b:0;i:15168;b:0;i:15174;b:0;i:15179;b:0;i:15191;b:0;i:15334;b:0;i:15622;b:0;i:15266;b:0;i:27632;b:1;i:15275;b:0;i:15332;b:0;i:15337;b:0;i:15303;b:0;i:15307;b:0;i:15336;b:0;i:15505;b:0;i:15444;b:0;i:15453;b:0;i:15529;b:0;i:15507;b:0;i:27640;b:1;i:18249;b:0;i:12612;b:0;i:13476;b:0;i:14636;b:0;i:15242;b:0;i:15648;b:0;i:15674;b:0;i:18449;b:0;i:15244;b:0;i:27645;b:1;i:27649;b:1;i:27650;b:0;i:27490;b:1;i:27626;b:1;i:27337;i:1;i:27525;i:1;i:27670;b:1;i:27666;b:1;i:27669;b:1;i:27676;b:1;i:27680;b:1;i:27685;b:1;i:27689;b:1;i:27478;b:1;i:27701;b:1;i:27716;b:1;i:27717;i:1;i:24316;b:1;i:23384;b:1;i:27737;b:1;i:27749;b:1;i:27755;b:1;i:27734;b:1;i:27754;b:1;i:27736;b:1;i:27779;b:1;i:27556;b:1;i:27784;b:1;i:27769;i:1;i:27696;b:1;i:27127;b:1;i:27113;b:1;i:27774;b:1;i:27806;b:1;i:26585;b:1;i:27616;b:1;i:26906;b:1;i:27786;b:1;i:27785;b:1;i:27835;i:1;i:27836;b:1;i:27841;b:1;i:27788;b:1;i:27739;b:1;i:27705;b:1;i:27857;b:1;i:27859;b:1;i:27874;b:1;i:27870;b:1;i:27858;b:1;i:27885;b:1;i:27783;b:1;i:24590;i:1;i:18524;i:0;i:21784;b:1;i:27897;b:1;i:22310;b:1;i:20619;i:0;i:27849;b:1;i:27765;b:1;i:27682;b:1;i:27635;b:1;i:27633;b:1;i:22278;b:1;i:20975;i:0;i:27898;b:1;i:27741;b:1;i:27916;b:1;i:27777;b:1;i:27923;b:1;i:27805;b:1;i:27930;b:1;i:27929;b:1;i:27062;b:1;i:27950;i:1;i:27752;b:1;i:27957;b:1;i:27960;b:1;i:27961;b:1;i:27876;b:1;i:27962;b:1;i:27844;b:1;i:27973;b:1;i:27975;b:1;i:27978;b:1;i:27971;b:1;i:27983;b:1;i:27990;b:1;i:27994;b:1;i:27995;b:1;i:27993;b:1;i:27988;b:1;i:27861;b:1;i:28003;b:1;i:27980;b:1;i:28002;b:1;i:27920;b:1;i:28033;b:1;i:28009;b:1;i:28008;b:1;i:28036;b:1;i:28034;b:1;i:28043;b:1;i:28053;b:1;i:28058;b:1;i:28038;b:1;i:28031;b:1;i:28073;b:1;i:28059;b:1;i:28047;b:1;i:28046;b:1;i:28054;b:1;i:28088;b:1;i:28090;b:1;i:28096;b:1;i:28099;b:1;i:27986;b:1;i:28108;b:1;i:28069;b:1;i:28113;b:1;i:26668;b:1;i:28125;b:1;i:28137;b:1;i:28072;b:1;i:28141;b:1;i:28142;b:1;i:28147;b:1;i:28149;b:1;i:28150;b:1;i:28152;b:1;i:28180;b:1;i:28183;b:1;i:28010;b:1;i:28203;b:1;i:28206;b:1;i:28205;b:1;i:28209;b:1;i:28032;b:1;i:28111;b:1;i:28202;b:1;i:28167;b:1;i:27938;b:1;i:27933;b:1;i:28158;b:1;i:10078;i:0;i:28181;b:1;i:28241;b:1;i:28249;b:1;i:28225;b:1;i:28193;b:1;i:28254;b:1;i:28261;b:1;i:28273;b:1;i:28216;b:1;i:28280;b:1;i:23669;i:1;i:28294;b:1;i:28251;b:1;i:28271;b:1;i:28296;b:1;i:28295;b:1;i:27794;b:1;i:28298;b:1;i:28307;b:1;i:28312;b:1;i:28317;b:1;i:28320;i:1;i:28323;b:1;i:28328;b:1;i:28329;b:1;i:28330;b:1;i:28338;b:1;i:28342;b:1;i:28343;b:1;i:28297;b:1;i:28354;b:1;i:28353;b:1;i:28374;b:1;i:28383;b:1;i:28384;b:1;i:28389;b:1;i:28404;b:1;i:28401;b:1;i:28407;b:1;i:27883;b:1;i:28420;b:1;i:28424;b:1;i:28335;i:1;i:28287;b:1;i:28285;b:1;i:28439;b:1;i:28390;b:1;i:28441;b:1;i:28381;b:1;i:28299;b:1;i:28446;b:1;i:28454;b:1;i:28475;b:1;i:28419;b:1;i:28518;b:1;i:28525;b:1;i:28463;b:1;i:28534;b:1;i:28536;b:1;i:28542;b:1;i:28509;b:1;i:28417;b:1;i:28494;b:1;i:28568;b:1;i:28570;b:1;i:28393;b:1;i:28582;b:1;i:28584;b:1;i:28589;b:1;i:28592;b:1;i:28593;b:1;i:27812;b:1;i:28606;b:1;i:28607;i:1;i:28617;b:1;i:28624;b:1;i:28625;b:1;i:28630;b:1;i:28644;b:1;i:28556;b:1;i:28569;b:1;i:28590;b:1;i:28627;b:1;i:28669;b:1;i:28413;b:1;i:28425;b:1;i:28612;b:1;i:28372;b:1;i:28676;b:1;i:28675;b:1;i:28674;b:1;i:28636;b:1;i:28681;b:1;i:28659;b:1;i:28696;b:1;i:28637;b:1;i:28640;b:1;i:28717;b:1;i:28657;b:1;i:28726;b:1;i:28013;b:1;i:28545;b:1;i:28739;b:1;i:28703;b:1;i:28749;b:1;i:28754;b:1;i:28765;b:1;i:28727;i:1;i:28787;b:1;i:28777;b:1;i:28766;b:1;i:28823;b:1;i:28822;b:1;i:28820;b:1;i:28826;i:1;i:28638;i:1;i:28725;b:1;i:28830;b:1;i:28776;b:1;i:28118;b:1;i:28742;b:1;i:28851;b:1;i:28857;b:1;i:28859;b:1;i:28876;i:1;i:28877;b:1;i:28763;b:1;i:28764;b:1;i:28889;b:1;i:28893;i:1;i:28291;b:1;i:28896;b:1;i:28795;b:1;i:28880;b:1;i:28895;b:1;i:28686;b:1;i:28714;b:1;i:28950;b:1;i:28785;b:1;i:28947;b:1;i:28953;b:1;i:28913;b:1;i:28961;b:1;i:28564;b:1;i:28775;b:1;i:28970;b:1;i:28986;b:1;i:28994;b:1;i:28829;b:1;i:28983;b:1;i:28869;b:1;i:29002;b:1;i:28946;b:1;i:29008;b:1;i:29007;b:1;i:29024;b:1;i:28996;b:1;i:29028;b:1;i:29043;b:1;i:28931;b:1;i:28911;b:1;i:28871;b:1;i:28808;b:1;i:29055;b:1;i:29053;b:1;i:29027;b:1;i:28918;b:1;i:28964;b:1;i:29070;b:1;i:29072;b:1;i:28998;i:1;i:28979;b:1;i:29084;b:1;i:29081;b:1;i:29082;b:1;i:29085;b:1;i:28867;b:1;i:29113;b:1;i:29094;b:1;i:29095;b:1;i:29133;b:1;i:29140;b:1;i:29149;b:1;i:29156;b:1;i:29166;b:1;i:29107;b:1;i:29112;b:1;i:29176;b:1;i:29154;b:1;i:29192;b:1;i:29195;b:1;i:29186;b:1;i:29165;b:1;i:29164;b:1;i:29197;b:1;i:29215;b:1;i:29216;b:1;i:29226;b:1;i:29228;b:1;i:29229;b:1;i:29233;b:1;i:29232;b:1;i:29230;b:1;i:29231;b:1;i:29205;b:0;i:29239;b:1;i:29253;b:1;i:29260;b:1;i:29263;b:1;i:27912;i:1;i:29266;i:1;i:29268;b:1;i:29270;b:1;i:29274;b:1;i:29190;b:1;i:29279;b:1;i:29243;b:1;i:29251;b:1;i:29041;b:1;i:28936;b:1;i:28846;b:1;i:28415;b:1;i:29304;b:1;i:29288;b:1;i:29303;b:0;i:29281;b:0;i:29291;b:0;i:29308;b:1;i:29311;i:1;i:29196;b:1;i:29250;b:1;i:29194;b:0;i:29199;b:1;i:29252;b:0;i:29283;b:1;i:29322;b:1;i:29327;b:1;i:29326;b:1;i:28467;b:1;i:29306;b:1;i:29052;b:1;i:29319;b:1;i:29335;b:1;i:29241;b:1;i:29059;b:1;i:29062;b:1;i:29096;b:1;i:29038;b:1;i:28736;b:1;i:29349;b:1;i:29350;b:1;i:28061;b:1;i:28782;b:1;i:28984;b:1;i:29381;b:1;i:29382;b:1;i:29369;b:1;i:29393;b:1;i:28786;b:1;i:29396;b:1;i:29405;b:1;i:29380;b:1;i:29414;b:1;i:29378;i:1;i:29377;i:1;i:29210;b:1;i:29376;i:1;i:29407;i:1;i:29397;i:1;i:29412;b:1;i:29421;b:1;i:29328;b:1;i:29320;b:1;i:29434;b:1;i:29240;b:1;i:29423;b:1;i:29468;b:1;i:29409;b:1;i:29445;b:1;i:27249;b:1;i:29479;b:1;i:29493;b:1;i:29497;b:1;i:29499;b:1;i:29503;b:1;i:29504;b:1;i:29501;b:0;i:29474;b:1;i:29510;b:1;i:29400;b:1;i:29394;b:1;i:29470;b:1;i:29529;b:1;i:29541;b:1;i:29544;b:1;i:29432;b:1;i:29404;b:1;i:29448;b:1;i:29505;b:1;i:29502;b:1;i:29570;b:1;i:29500;b:1;i:29580;b:1;i:29579;b:1;i:29559;b:1;i:29557;b:1;i:29454;b:1;i:29457;b:1;i:29588;b:1;i:29595;b:1;i:29422;b:1;i:29221;b:1;i:29553;b:1;i:29616;b:1;i:29618;b:1;i:29391;b:1;i:29620;b:1;i:29615;b:1;i:29550;b:1;i:29629;b:1;i:29646;i:1;i:29639;b:1;i:29655;b:1;i:29660;b:1;i:29667;b:1;i:29619;b:1;i:29668;b:1;i:29638;b:1;i:29645;b:1;i:29677;b:1;i:29657;b:1;i:29640;b:1;i:29631;b:1;i:29696;b:1;i:29698;b:1;i:29702;b:1;i:29706;b:1;i:29330;b:1;i:29717;b:1;i:29725;b:1;i:29608;b:1;i:29606;b:1;i:29762;b:1;i:29770;b:1;i:29780;b:1;i:29782;b:1;i:29785;b:1;i:29795;b:1;i:29539;b:1;i:29796;b:1;i:29742;b:1;i:29791;b:1;i:29810;b:1;i:29800;b:1;i:29822;b:1;i:29827;b:1;i:29705;b:1;i:29843;b:1;i:29841;b:1;i:29856;b:1;i:29833;b:1;i:29769;b:1;i:29819;b:1;i:29883;b:1;i:29880;b:1;i:29888;b:1;i:29887;b:1;i:29768;b:1;i:29900;b:1;i:29920;b:1;i:29272;b:1;i:29914;b:1;i:29899;b:1;i:29834;b:1;i:29831;b:1;i:29924;b:1;i:29783;b:1;i:29937;b:1;i:29790;b:1;i:29911;b:1;i:29946;b:1;i:29958;b:1;i:29957;b:1;i:29955;b:1;i:29861;b:1;i:29919;b:1;i:29968;b:1;i:29980;b:1;i:29871;b:1;i:29989;b:1;i:29994;b:1;i:29879;b:1;i:30007;b:1;i:29959;b:1;i:30031;b:1;i:30037;b:1;i:30041;b:1;i:30053;b:1;i:29891;b:1;i:30050;b:1;i:30056;b:1;i:30058;b:1;i:30055;b:1;i:30080;b:1;i:30071;b:1;i:30085;b:1;i:30075;b:1;i:30097;b:1;i:30098;b:0;i:29703;b:1;i:30119;b:1;i:29965;b:1;i:30038;b:1;i:30136;b:1;i:29987;b:1;i:30121;b:1;i:30145;b:1;i:30032;b:1;i:30153;b:1;i:30165;b:1;i:30164;b:1;i:30167;b:1;i:30168;b:1;i:30184;b:1;i:30133;b:1;i:30200;b:1;i:30180;b:1;i:30059;b:1;i:30060;b:1;i:30115;b:1;i:30205;b:1;i:30127;b:1;i:30159;b:1;i:30161;b:1;i:30221;b:1;i:30237;b:1;i:30236;b:1;i:30244;b:1;i:30151;b:1;i:30246;b:1;i:30253;b:1;i:30257;b:1;i:30245;b:1;i:30273;b:1;i:30278;b:1;i:30274;b:1;i:30282;b:1;i:30284;b:1;i:30285;b:1;i:30131;b:1;i:30254;b:1;i:30290;b:1;i:30288;b:1;i:30293;b:1;i:30294;b:1;i:30276;b:1;i:30308;b:1;i:30310;b:1;i:30312;b:1;i:30331;b:1;i:30283;b:1;i:30342;b:1;i:30344;b:1;i:30343;b:1;i:30345;b:1;i:30334;b:1;i:30327;b:1;i:30363;b:1;i:30324;b:1;i:30371;b:1;i:30260;b:1;i:30256;b:1;i:30375;b:1;i:30364;b:1;i:30357;b:1;i:30390;b:1;i:30391;b:1;i:30388;b:1;i:30396;b:1;i:30416;b:1;i:30415;b:1;i:30423;b:1;i:30421;b:1;i:30425;b:1;i:30432;b:1;i:30424;b:1;i:30438;b:1;i:30442;b:1;i:30446;b:1;i:30441;b:1;i:30422;b:1;i:30449;b:1;i:30369;b:1;i:30440;b:1;i:30455;b:1;i:30361;b:1;i:30387;b:1;i:30420;b:1;i:30476;b:1;i:30485;b:1;i:30355;b:1;i:30494;b:1;i:30490;b:1;i:30482;b:1;i:30510;b:1;i:30535;b:1;i:30504;b:1;i:30501;b:1;i:30540;b:1;i:30517;b:1;i:30303;b:1;i:30550;b:1;i:30553;b:1;i:30555;b:1;i:30459;b:1;i:30533;b:1;i:30521;b:1;i:30560;b:1;i:30532;b:1;i:30562;b:1;i:30575;b:1;i:30588;b:1;i:30598;b:1;i:30539;b:1;i:30618;b:1;i:30631;b:1;i:30630;b:1;i:30629;b:1;i:30599;b:1;i:30636;b:1;i:30624;b:1;i:30514;b:1;i:30655;i:0;i:30659;b:1;i:30663;b:1;i:30635;b:1;i:30513;b:1;i:30667;b:1;i:30665;b:1;i:30669;b:1;i:30684;b:1;i:30683;b:1;i:30691;b:1;i:30156;b:1;i:30395;b:1;i:30394;b:1;i:30696;b:1;i:30621;b:1;i:30451;b:1;i:30554;b:1;i:30681;b:1;i:30692;b:1;i:30720;b:1;i:30722;b:1;i:30695;b:1;i:30714;b:1;i:30728;b:1;i:30730;b:1;i:30702;b:1;i:30563;b:1;i:30745;b:1;i:30687;b:1;i:30751;b:1;i:30759;b:1;i:30754;b:1;i:30768;b:1;i:30769;b:1;i:30749;b:1;i:30770;b:1;i:30771;b:0;i:30780;b:1;i:30781;b:1;i:30785;b:1;i:30774;b:1;i:30807;b:1;i:30676;b:1;i:30823;b:1;i:30822;b:1;i:30740;b:1;i:30828;b:1;i:30703;b:1;i:30857;b:1;i:30811;b:1;i:30778;b:1;i:30777;b:1;i:30868;b:1;i:30878;b:1;i:30820;b:1;i:30813;b:1;i:30895;b:1;i:30799;b:1;i:30830;b:1;i:30626;b:1;i:30767;b:1;i:30904;b:1;i:30887;b:1;i:30845;b:1;i:30881;b:1;i:30743;b:1;i:30797;b:1;i:30925;b:1;i:30795;b:1;i:30872;b:1;i:30943;b:1;i:30929;b:1;i:30625;b:1;i:30628;b:1;i:30966;b:1;i:30902;b:1;i:30907;b:1;i:30899;b:1;i:30764;b:1;i:30956;b:1;i:30914;b:1;i:30809;b:1;i:30713;b:1;i:30978;b:1;i:31016;b:1;i:31031;b:1;i:30913;b:1;i:30718;b:1;i:30871;b:1;i:31020;b:1;i:31057;b:1;i:31021;b:1;i:31064;b:1;i:31071;b:1;i:30582;b:1;i:31030;b:1;i:31092;b:1;i:31005;b:1;i:30994;b:1;i:31103;b:1;i:31088;i:1;i:31115;b:1;i:31116;b:1;i:31089;b:1;i:31101;b:1;i:31078;b:1;i:31121;b:1;i:31122;b:1;i:31048;b:1;i:31130;b:1;i:30867;b:1;i:31110;b:1;i:31139;b:1;i:31141;b:1;i:31114;b:1;i:31158;b:1;i:31162;b:1;i:31001;b:1;i:31152;b:1;i:31156;b:1;i:31204;b:1;i:31153;b:1;i:31206;b:1;i:31056;b:1;i:31054;b:1;i:31211;b:1;i:31193;b:1;i:30627;b:1;i:31143;b:1;i:31099;b:1;i:31232;b:1;i:31216;b:1;i:31239;b:1;i:31258;b:1;i:31265;b:1;i:31218;b:1;i:31262;b:1;i:31252;b:1;i:31276;b:1;i:31278;i:1;i:31281;i:1;i:31293;b:1;i:31300;b:1;i:31221;b:1;i:31306;b:1;i:31310;b:1;i:31313;b:1;i:31309;b:1;i:31311;b:1;i:31312;b:1;i:31308;b:1;i:31226;b:1;i:31333;b:1;i:31334;b:1;i:31343;b:1;i:31346;b:1;i:31348;b:1;i:31355;b:1;i:31340;b:1;i:31360;b:1;i:31339;b:1;i:31361;b:1;i:31337;b:1;i:31304;b:1;i:31368;b:1;i:31370;b:1;i:31369;b:1;i:31380;b:1;i:31383;b:1;i:31389;b:1;i:31374;b:1;i:31397;b:1;i:31396;b:1;i:31292;b:1;i:31406;b:1;i:31420;b:1;i:31436;b:1;i:31437;b:1;i:31390;b:1;i:31442;b:1;i:31448;b:1;i:31449;b:1;i:31450;b:1;i:31433;b:1;i:31438;b:1;i:31451;b:1;i:31458;b:1;i:31461;b:1;i:31460;b:1;i:31473;b:1;i:31477;b:1;i:31476;b:1;i:31425;i:1;i:31462;b:1;i:31488;b:1;i:31493;b:1;i:31490;b:1;i:31513;b:1;i:31492;b:1;i:31516;b:1;i:31536;b:1;i:31533;b:1;i:31419;b:1;i:31541;b:1;i:31547;b:1;i:31520;b:1;i:31558;b:1;i:31557;b:1;i:31553;b:1;i:31517;b:1;i:31527;b:1;i:31526;b:1;i:31572;b:1;i:31567;b:1;i:31566;b:1;i:31577;b:1;i:31382;b:1;i:31582;b:1;i:31590;b:1;i:31592;b:1;i:31594;b:1;i:31591;b:1;i:31599;i:1;i:31600;b:1;i:31509;b:1;i:31612;b:1;i:31617;b:1;i:31622;b:1;i:31583;b:1;i:31635;b:1;i:30862;b:1;i:31657;b:1;i:31660;b:1;i:31587;b:1;i:31664;b:1;i:31674;b:1;i:31649;b:1;i:31040;b:1;i:31626;b:1;i:31624;b:1;i:31681;i:1;i:31384;b:1;i:31689;b:1;i:31316;b:1;i:31619;b:1;i:31564;b:1;i:31463;b:1;i:31683;b:1;i:31629;b:1;i:31714;b:1;i:31716;b:1;i:31720;b:1;i:31722;b:1;i:31718;b:1;i:31731;b:1;i:31738;b:1;i:31736;b:1;i:31737;b:1;i:31739;b:1;i:31639;b:1;i:31756;b:1;i:31743;b:1;i:31745;b:1;i:31762;b:1;i:31763;b:1;i:31740;b:1;i:31766;b:1;i:31765;b:1;i:31801;b:1;i:31813;b:1;i:31815;b:1;i:31817;b:1;i:31825;b:1;i:31824;b:1;i:31826;b:1;i:31829;b:1;i:31834;b:1;i:31846;b:1;i:31836;b:1;i:31869;b:1;i:31868;b:1;i:31872;b:1;i:31827;b:1;i:31890;b:1;i:31875;b:1;i:31887;b:1;i:31823;b:1;i:31897;b:1;i:31900;b:1;i:31907;b:1;i:31908;b:1;i:31926;b:1;i:31927;i:1;i:31862;b:1;i:31804;b:1;i:31949;b:1;i:31574;b:1;i:31981;b:1;i:31803;b:1;i:31992;b:1;i:31996;b:1;i:31998;b:1;i:32000;b:1;i:32007;i:1;i:32010;b:1;i:31995;b:1;i:32028;b:1;i:32031;i:1;i:32038;b:1;i:32040;b:1;i:32041;b:1;i:32037;b:1;i:32051;b:1;i:32060;b:1;i:32058;b:1;i:31963;b:1;i:32063;b:1;i:31952;b:1;i:32048;b:1;i:32075;b:1;i:32077;b:1;i:32074;b:1;i:32076;b:1;i:32083;b:1;i:32086;b:1;i:32089;b:1;i:32088;b:1;i:32096;b:1;i:32098;b:1;i:32073;b:1;i:32099;b:1;i:32115;b:1;i:32120;b:1;i:32124;b:1;i:32123;b:1;i:32121;b:1;i:32114;b:1;i:32127;b:1;i:32125;b:1;i:32139;b:1;i:32146;b:1;i:32150;i:1;i:32145;b:1;i:32167;b:1;i:32169;b:1;i:32117;b:1;i:32184;b:1;i:32206;b:1;i:32211;b:1;i:32223;b:1;i:32224;b:1;i:32242;b:1;i:32249;b:1;i:32258;b:1;i:32255;b:1;i:32254;b:1;i:32259;b:1;i:32253;b:1;i:32260;b:1;i:32262;b:1;i:32265;b:1;i:31984;b:1;i:32273;b:1;i:32221;b:1;i:32290;b:1;i:32202;b:1;i:32187;b:1;i:32143;b:1;i:32243;b:1;i:32310;b:1;i:32313;b:1;i:32218;b:1;i:32321;b:1;i:32328;b:1;i:32340;b:1;i:32334;i:1;i:32330;b:1;i:32356;b:1;i:32365;b:1;i:32298;b:1;i:32363;b:1;i:32367;b:1;i:32362;b:1;i:32327;b:1;i:32376;b:1;i:32369;b:1;i:32378;b:1;i:32381;b:1;i:32379;b:1;i:32384;b:1;i:32397;b:1;i:32396;b:1;i:32400;b:1;i:32399;b:1;i:32413;i:1;i:32394;b:1;i:32401;b:1;i:32443;b:1;i:32385;b:1;i:32294;b:1;i:32370;b:1;i:32212;b:1;i:32468;b:1;i:32319;b:1;i:32455;b:1;i:32458;b:1;i:32481;b:1;i:32493;b:1;i:32495;b:1;i:32157;b:1;i:32513;b:1;i:32515;b:1;i:32496;b:1;i:32526;i:1;i:32539;b:1;i:32549;b:1;i:32490;b:1;i:32554;b:1;i:32581;b:1;i:32602;b:1;i:32604;b:1;i:32508;b:1;i:32506;b:1;i:32626;b:1;i:32600;b:1;i:32638;b:1;i:32065;b:1;i:32577;b:1;i:32653;b:1;i:32667;b:1;i:32668;b:1;i:32664;b:1;i:32662;b:1;i:32701;b:1;i:32712;b:1;i:32718;b:1;i:32719;b:1;i:32721;b:1;i:32693;b:1;i:32726;b:1;i:32731;b:1;i:32734;b:1;i:32735;b:1;i:32717;b:1;i:32752;b:1;i:32753;b:1;i:32755;b:1;i:32741;b:1;i:32479;b:1;i:32758;b:1;i:32764;i:1;i:32637;b:1;i:32767;b:1;i:32675;b:1;i:32787;b:1;i:32784;b:1;i:32749;b:1;i:32815;b:1;i:32798;b:1;i:32805;b:1;i:32809;b:1;i:32807;b:1;i:32828;b:1;i:32644;b:1;i:32835;b:1;i:32839;b:1;i:32836;b:1;i:32838;b:1;i:32800;b:1;i:32850;b:1;i:32837;b:1;i:32858;b:1;i:32859;b:1;i:32856;b:1;i:32865;b:1;i:32868;b:1;i:32870;b:1;i:32832;b:1;i:32869;b:1;i:32874;b:1;i:32462;b:1;i:32898;b:1;i:32901;b:1;i:32904;b:1;i:32876;b:1;i:32908;b:1;i:32853;b:1;i:32852;b:1;i:32907;b:1;i:32911;b:1;i:32524;b:1;i:32759;b:1;i:17543;b:1;i:32930;b:1;i:32933;b:1;i:32932;b:1;i:32936;b:1;i:32941;b:1;i:32944;b:1;i:32945;b:1;i:32872;b:1;i:32969;b:1;i:32974;b:1;i:32978;b:1;i:32994;b:1;i:32973;b:1;i:32970;b:1;i:33010;b:1;i:33008;b:1;i:32993;b:1;i:32954;b:1;i:33017;b:1;i:32990;b:1;i:32982;b:1;i:32946;b:1;i:33034;b:1;i:33048;b:1;i:33055;b:1;i:33062;b:1;i:32877;b:1;i:33067;b:1;i:33077;b:1;i:33001;b:1;i:32818;b:1;i:33087;b:1;i:32766;i:1;i:33061;b:1;i:33091;b:1;i:33094;b:1;i:33030;i:1;i:33114;b:1;i:33111;b:1;i:33086;b:1;i:33118;b:1;i:33115;b:1;i:33097;b:1;i:33129;b:1;i:33133;b:1;i:33136;b:1;i:33143;b:1;i:33148;b:1;i:33141;b:1;i:33063;b:1;i:33158;b:1;i:33159;b:1;i:33099;b:1;i:32964;b:1;i:33191;b:1;i:33195;b:1;i:33196;b:1;i:33194;b:1;i:33179;b:1;i:33207;b:1;i:33211;b:1;i:33219;b:1;i:33090;b:1;i:33058;b:1;i:33226;b:1;i:33228;b:1;i:33103;b:1;i:33229;b:1;i:33209;b:1;i:33232;b:1;i:33079;b:1;i:33238;b:1;i:33242;b:1;i:33244;b:1;i:33246;b:1;i:33256;b:1;i:33258;b:1;i:33235;b:1;i:33183;b:1;i:33260;b:1;i:33262;b:1;i:33264;b:1;i:33265;b:1;i:33266;b:1;i:33210;i:1;i:33224;b:1;i:33227;b:1;i:33285;b:1;i:33289;b:1;i:33286;b:1;i:33290;b:1;i:33218;b:1;i:33307;b:1;i:32997;b:1;i:33325;b:1;i:33250;b:1;i:33323;b:1;i:33026;b:1;i:33050;b:1;i:33301;b:1;i:33300;b:1;i:32931;b:1;i:33046;b:1;i:33344;b:1;i:33351;b:1;i:33355;b:1;i:33360;b:1;i:33120;b:1;i:33373;b:1;i:33376;b:1;i:33147;b:1;i:33382;b:1;i:33394;b:1;i:33174;b:1;i:33308;b:1;i:33406;b:1;i:33409;b:1;i:33386;b:1;i:33410;b:1;i:33438;b:1;i:33395;b:1;i:33124;b:1;i:33451;b:1;i:33448;b:1;i:33442;b:1;i:33456;b:1;i:32899;b:1;i:33465;b:1;i:33466;b:1;i:33311;b:1;i:33476;b:1;i:33480;b:1;i:33484;b:1;i:33368;b:1;i:33324;b:1;i:33453;b:1;i:33407;b:1;i:33487;b:1;i:33469;b:1;i:33493;b:1;i:33421;b:1;i:33379;b:1;i:33497;b:1;i:33486;b:1;i:33483;b:1;i:33499;b:1;i:33412;b:1;i:33502;b:1;i:33295;b:1;i:33501;b:1;i:33515;b:1;i:33492;b:1;i:33454;b:1;i:33525;b:1;i:33520;b:1;i:33532;b:1;i:33507;b:1;i:33510;b:1;i:33543;b:1;i:33547;b:1;i:33318;b:1;i:33556;b:1;i:33518;b:1;i:33517;b:1;i:33550;b:1;i:33560;b:1;i:33566;b:1;i:33297;b:1;i:33562;b:1;i:33571;b:1;i:33559;b:1;i:33578;b:1;i:33579;b:1;i:33580;b:1;i:33588;b:1;i:33587;b:1;i:33598;b:1;i:33553;b:1;i:33609;b:1;i:33612;b:1;i:33619;b:1;i:33611;b:1;i:33589;b:1;i:33540;b:1;i:33625;b:1;i:33627;b:1;i:33629;b:1;i:33628;b:1;i:33630;b:1;i:33638;b:1;i:33640;b:1;i:33488;b:1;i:33621;b:1;i:33643;b:1;i:33617;b:1;i:33652;b:1;i:33633;b:1;i:33657;b:1;i:33653;b:1;i:33663;b:1;i:33666;b:1;i:28191;i:1;i:33673;b:1;i:33678;b:1;i:33670;b:1;i:33682;b:1;i:33669;b:1;i:33685;b:1;i:33651;b:1;i:33702;b:1;i:33698;b:1;i:33679;b:1;i:33655;b:1;i:33712;b:1;i:33714;b:1;i:33713;b:1;i:33701;b:1;i:33739;b:1;i:33733;b:1;i:33742;b:1;i:33622;b:1;i:33745;b:1;i:33746;b:1;i:33744;b:1;i:33737;b:1;i:33719;b:1;i:33687;b:1;i:33684;b:1;i:33773;b:1;i:33774;b:1;i:33778;b:1;i:33783;b:1;i:33786;b:1;i:33637;b:1;i:33671;b:1;i:33727;b:1;i:33730;b:1;i:33798;b:1;i:33799;b:1;i:33763;b:1;i:33781;b:1;i:33815;b:1;i:33827;b:1;i:33755;b:1;i:33810;b:1;i:33845;b:1;i:33847;b:1;i:33776;b:1;i:33853;b:1;i:33859;b:1;i:33836;b:1;i:33860;b:1;i:33877;b:1;i:33883;b:1;i:33867;b:1;i:33834;b:1;i:33835;b:1;i:33870;b:1;i:33903;b:1;i:33904;b:1;i:33839;b:1;i:33848;b:1;i:33820;b:1;i:33814;b:1;i:33858;b:1;i:33826;b:1;i:33927;b:1;i:33837;b:1;i:33911;b:1;i:33930;b:1;i:33817;b:1;i:33936;b:1;i:33942;b:1;i:33958;b:1;i:33953;b:1;i:33887;b:1;i:33896;b:1;i:33971;b:1;i:33974;b:1;i:33987;b:1;i:33986;b:1;i:33989;b:1;i:33941;b:1;i:33992;b:1;i:34004;b:1;i:33938;b:1;i:34018;b:1;i:34020;b:1;i:34019;b:1;i:34008;b:1;i:34009;b:1;i:34011;b:1;i:34046;b:1;i:33998;b:1;i:34060;b:1;i:34062;b:1;i:34036;b:1;i:33975;b:1;i:34082;b:1;i:34094;b:1;i:34086;b:1;i:34098;b:1;i:34002;b:1;i:34114;b:1;i:34123;b:1;i:34116;b:1;i:34139;b:1;i:34142;b:1;i:34143;b:1;i:34105;b:1;i:34149;b:1;i:34151;b:1;i:34156;b:1;i:34136;b:1;i:34133;b:1;i:34118;b:1;i:34157;b:1;i:34161;b:1;i:34160;b:1;i:34103;b:1;i:33855;b:1;i:34171;b:1;i:34147;b:1;i:34180;b:1;i:34124;b:1;i:34138;b:1;i:34184;b:1;i:34100;b:1;i:34177;b:1;i:34155;b:1;i:34195;b:1;i:34204;b:1;i:34218;b:1;i:34168;b:1;i:34097;b:1;i:34229;b:1;i:34233;b:1;i:34166;b:1;i:34241;b:1;i:34249;b:1;i:34221;b:1;i:34252;b:1;i:34251;b:1;i:34270;b:1;i:34279;b:1;i:34269;b:1;i:34267;b:1;i:34293;b:1;i:34273;b:1;i:34272;b:1;i:34305;b:1;i:34303;b:1;i:34289;b:1;i:34264;b:1;i:34304;b:1;i:34325;b:1;i:34324;b:1;i:34326;b:1;i:34327;b:1;i:34334;b:1;i:34342;b:1;i:34290;b:1;i:34256;b:1;i:34345;b:1;i:34281;b:1;i:34319;b:1;i:34294;b:1;i:34352;b:1;i:34353;b:1;i:34261;b:1;i:34292;b:1;i:34349;b:1;i:34372;b:1;i:34378;b:1;i:34399;b:1;i:34398;b:1;i:34401;b:1;i:34404;b:1;i:34356;b:1;i:34299;b:1;i:34422;b:1;i:34427;b:1;i:34430;b:1;i:34418;b:1;i:34403;b:1;i:34432;b:1;i:34433;b:1;i:34442;b:1;i:34436;b:1;i:34451;b:1;i:34307;b:1;i:34439;b:1;i:34339;b:1;i:34425;b:1;i:34446;b:1;i:34490;b:1;i:34456;b:1;i:34463;b:1;i:34462;b:1;i:34500;b:1;i:34278;b:1;i:34510;b:1;i:34506;b:1;i:34513;b:1;i:34514;b:1;i:34478;b:1;i:34519;b:1;i:34520;b:1;i:34525;b:1;i:34526;b:1;i:34528;b:1;i:34450;b:1;i:34438;b:1;i:34529;b:1;i:34533;b:1;i:34540;b:1;i:34538;b:1;i:34539;b:1;i:34475;b:1;i:34549;b:1;i:34551;b:1;i:34480;b:1;i:34535;b:1;i:34515;b:1;i:34331;b:1;i:34562;b:1;i:34566;b:1;i:34570;b:1;i:34574;b:1;i:34429;b:1;i:34320;b:1;i:34585;b:1;i:34559;b:1;i:34565;b:1;i:34554;b:1;i:34613;b:1;i:34618;b:1;i:34553;b:1;i:34620;b:1;i:34621;b:1;i:34616;b:1;i:34632;b:1;i:34633;b:1;i:34589;b:1;i:34373;b:1;i:34648;b:1;i:34655;b:1;i:34659;b:1;i:34483;b:1;i:34614;b:1;i:34675;b:1;i:34671;b:1;i:34629;b:1;i:34676;b:1;i:34643;b:1;i:34682;b:1;i:34670;b:1;i:34639;b:1;i:34602;b:1;i:34617;b:1;i:34581;b:1;i:34699;b:1;i:34707;b:1;i:34708;b:1;i:34509;b:1;i:34688;b:1;i:34725;b:1;i:34569;b:1;i:34736;b:1;i:34737;b:1;i:34738;b:1;i:34598;b:1;i:33933;i:1;i:34579;i:1;i:34612;i:1;i:34537;b:1;i:34609;b:1;i:34744;b:1;i:34722;b:1;i:34760;b:1;i:34761;b:1;i:34762;b:1;i:34766;b:1;i:34732;b:1;i:34769;b:1;i:34768;b:1;i:34759;b:1;i:34631;b:1;i:34441;b:1;i:34774;b:1;i:34773;b:1;i:34782;b:1;i:34750;b:1;i:34544;b:1;i:34815;b:1;i:34819;b:1;i:34828;b:1;i:34808;b:1;i:34765;b:1;i:34850;b:1;i:34816;b:1;i:34831;b:1;i:34830;b:1;i:34829;b:1;i:34827;b:1;i:34837;b:1;i:34878;b:1;i:34887;b:1;i:34894;b:1;i:34888;b:1;i:34881;b:1;i:34794;b:1;i:34910;b:1;i:34895;b:1;i:34916;b:1;i:34920;b:1;i:34917;b:1;i:34923;b:1;i:34919;b:1;i:34929;b:1;i:34934;b:1;i:34857;b:1;i:34905;b:1;i:34904;b:1;i:34950;b:1;i:34930;b:1;i:34844;b:1;i:34958;b:1;i:34959;b:1;i:34960;b:1;i:34927;b:1;i:34966;b:1;i:34967;b:1;i:34961;b:1;i:34971;b:1;i:34953;b:1;i:34972;b:1;i:34957;b:1;i:34973;b:1;i:34969;b:1;i:34886;b:1;i:34059;b:1;i:34965;b:1;i:34999;b:1;i:35009;b:1;i:35003;b:1;i:34861;b:1;i:34790;b:1;i:35012;b:1;i:35014;b:1;i:34995;b:1;i:35018;b:1;i:35027;b:1;i:35032;b:1;i:35024;b:1;i:34944;b:1;i:35042;b:1;i:35029;b:1;i:35043;b:1;i:35046;b:1;i:35047;b:1;i:34843;b:1;i:34896;b:1;i:35040;b:1;i:35057;b:1;i:35074;b:1;i:35071;b:1;i:35076;b:1;i:34237;b:1;i:34954;b:1;i:35082;b:1;i:35089;b:1;i:35101;b:1;i:35069;b:1;i:34680;b:1;i:35103;b:1;i:35115;b:1;i:35120;b:1;i:35121;b:1;i:35117;b:1;i:35116;b:1;i:35122;b:1;i:35041;b:1;i:35119;b:1;i:35083;b:1;i:35090;b:1;i:35037;b:1;i:35062;b:1;i:35060;b:1;i:35056;b:1;i:35100;b:1;i:35169;b:1;i:35087;b:1;i:35127;b:1;i:35184;b:1;i:35188;b:1;i:35171;b:1;i:35172;b:1;i:35173;b:1;i:35190;b:1;i:35191;b:1;i:35194;b:1;i:35201;b:1;i:35193;b:1;i:35197;b:1;i:35198;b:1;i:35162;b:1;i:35177;b:1;i:35199;b:1;i:35221;b:1;i:35220;b:1;i:35218;b:1;i:35225;b:1;i:35196;b:1;i:35232;b:1;i:35238;b:1;i:35226;b:1;i:35144;b:1;i:35254;b:1;i:35253;b:1;i:35256;b:1;i:35257;b:1;i:35255;b:1;i:35156;b:1;i:35265;b:1;i:35267;b:1;i:35167;b:1;i:35272;b:1;i:35271;b:1;i:35211;b:1;i:35289;b:1;i:35291;b:1;i:17310;i:0;i:35307;b:1;i:35317;b:1;i:35278;b:1;i:35136;b:1;i:35269;b:1;i:35327;b:1;i:35202;b:1;i:35333;b:1;i:35336;b:1;i:35339;b:1;i:35354;b:1;i:35329;b:1;i:35356;b:1;i:35361;b:1;i:35367;b:1;i:35131;b:1;i:35369;b:1;i:35379;b:1;i:35371;b:1;i:35396;b:1;i:35395;b:1;i:35402;b:1;i:35424;b:1;i:35434;b:1;i:35428;b:1;i:35436;b:1;i:35438;b:1;i:35433;b:1;i:35445;b:1;i:35388;b:1;i:35450;b:1;i:35453;b:1;i:35422;b:1;i:35423;b:1;i:35439;b:1;i:35334;b:1;i:35467;b:1;i:35469;b:1;i:35471;b:1;i:35478;b:1;i:35437;b:1;i:35494;b:1;i:35497;b:1;i:35468;b:1;i:35320;b:1;i:35500;b:1;i:35297;b:1;i:35504;b:1;i:35507;b:1;i:33028;i:1;i:35513;b:1;i:35417;b:1;i:35511;b:1;i:35451;b:1;i:35488;b:1;i:35541;b:1;i:35545;b:1;i:35539;b:1;i:35547;b:1;i:35540;b:1;i:35548;b:1;i:35551;b:1;i:35525;b:1;i:35556;b:1;i:35557;b:1;i:35531;b:1;i:35449;b:1;i:35554;b:1;i:35586;b:1;i:35549;b:1;i:35495;b:1;i:35597;b:1;i:35508;b:1;i:35414;b:1;i:35603;b:1;i:35628;b:1;i:35621;b:1;i:35634;b:1;i:35636;b:1;i:35490;b:1;i:35645;b:1;i:35644;b:1;i:35648;b:1;i:35640;b:1;i:35650;b:1;i:35535;b:1;i:35601;b:1;i:35655;b:1;i:35627;b:1;i:35602;b:1;i:35658;b:1;i:35637;b:1;i:35665;b:1;i:35672;b:1;i:35676;b:1;i:35669;b:1;i:35674;b:1;i:35679;b:1;i:35613;b:1;i:35564;b:1;i:35565;b:1;i:35690;b:1;i:35689;b:1;i:35693;b:1;i:35709;b:1;i:35533;b:1;i:35615;b:1;i:35593;b:1;i:35698;b:1;i:35697;b:1;i:35710;b:1;i:35578;b:1;i:35631;b:1;i:35716;b:1;i:35723;b:1;i:35728;b:1;i:35731;b:1;i:35704;b:1;i:35730;b:1;i:35726;b:1;i:35738;b:1;i:35745;b:1;i:35574;b:1;i:35758;b:1;i:35737;b:1;i:35767;b:1;i:35768;b:1;i:35619;b:1;i:35717;b:1;i:35774;b:1;i:35775;b:1;i:35677;b:1;i:35779;b:1;i:35778;b:1;i:35780;b:1;i:35735;b:1;i:35694;b:1;i:35787;b:1;i:35801;b:1;i:35807;b:1;i:35812;b:1;i:35811;b:1;i:35741;b:1;i:35840;b:1;i:35842;b:1;i:35847;b:1;i:35839;b:1;i:35849;b:1;i:35850;b:1;i:35851;b:1;i:35819;i:1;i:35855;b:1;i:35748;b:1;i:35739;b:1;i:35865;b:1;i:35876;b:1;i:35877;b:1;i:35881;b:1;i:35888;b:1;i:35889;b:1;i:35890;b:1;i:35838;b:1;i:35895;b:1;i:35771;b:1;i:35893;b:1;i:35798;i:1;i:35864;i:1;i:35907;b:1;i:35906;i:1;i:35875;b:1;i:35873;b:1;i:35897;b:1;i:35925;b:1;i:35885;b:1;i:35930;i:1;i:35935;b:1;i:35943;b:1;i:35938;b:1;i:35843;b:1;i:35947;b:1;i:35961;b:1;i:35909;i:1;i:35884;b:1;i:35939;b:1;i:35967;b:1;i:35972;b:1;i:35929;b:1;i:35950;b:1;i:35983;b:1;i:35858;b:1;i:35974;b:1;i:35923;b:1;i:36005;b:1;i:36007;b:1;i:35996;b:1;i:36009;b:1;i:36016;b:1;i:36013;b:1;i:36024;b:1;i:35988;b:1;i:36028;b:1;i:36000;b:1;i:36038;b:1;i:36046;b:1;i:36043;b:1;i:36049;b:1;i:36041;b:1;i:35952;b:1;i:36031;b:1;i:35946;b:1;i:36034;b:1;i:35975;b:1;i:35973;i:1;i:35879;b:1;i:35977;b:1;i:36067;b:1;i:36065;b:1;i:36072;b:1;i:36071;b:1;i:35792;i:1;i:35760;i:1;i:35757;i:1;i:36076;b:1;i:36085;b:1;i:36087;b:1;i:36086;b:1;i:36064;b:1;i:36103;b:1;i:36081;b:1;i:36105;b:1;i:36101;b:1;i:36107;b:1;i:36091;b:1;i:36106;b:1;i:36128;b:1;i:36117;i:1;i:36141;b:1;i:36136;b:1;i:36144;b:1;i:36143;b:1;i:36145;b:1;i:36068;b:1;i:36147;b:1;i:36149;b:1;i:36151;i:1;i:36152;b:1;i:36104;b:1;i:35954;b:1;i:36160;b:1;i:36164;b:1;i:36167;b:1;i:36166;b:1;i:36170;b:1;i:36123;b:1;i:36180;b:1;i:36181;b:1;i:36184;b:1;i:36187;b:1;i:36169;b:1;i:36193;b:1;i:36096;b:1;i:36197;b:1;i:36191;b:1;i:35773;b:1;i:36208;b:1;i:36173;b:1;i:36223;i:1;i:36232;b:1;i:36178;b:1;i:36233;b:1;i:36234;b:1;i:36134;b:1;i:36074;b:1;i:36240;b:1;i:36250;b:1;i:36109;b:1;i:36228;i:1;i:36277;i:1;i:36023;b:1;i:36153;b:1;i:36256;b:1;i:36295;b:1;i:36275;b:1;i:36200;b:1;i:36201;b:1;i:36323;b:1;i:36305;b:1;i:36319;b:1;i:36226;b:1;i:36337;b:1;i:36338;b:1;i:36343;b:1;i:36342;i:1;i:36345;b:1;i:36335;b:1;i:36334;b:1;i:36352;b:1;i:36339;b:1;i:36369;i:1;i:36374;b:1;i:36253;b:1;i:36385;b:1;i:36389;b:1;i:36398;i:1;i:36301;b:1;i:35998;b:1;i:36386;b:1;i:36420;b:1;i:36425;b:1;i:36434;b:1;i:36348;b:1;i:36424;b:1;i:36364;b:1;i:36437;b:1;i:36259;b:1;i:36316;b:1;i:36415;b:1;i:36447;i:1;i:35871;b:1;i:36463;b:1;i:36428;b:1;i:36438;b:1;i:36468;b:1;i:36480;b:1;i:36478;b:1;i:36479;b:1;i:36484;b:1;i:36486;b:1;i:36472;b:1;i:36473;b:1;i:36391;b:1;i:36433;b:1;i:36430;b:1;i:36474;b:1;i:36503;b:1;i:36513;b:1;i:36477;b:1;i:36512;b:1;i:36516;b:1;i:36524;b:1;i:36525;b:1;i:36384;b:1;i:36414;b:1;i:36411;b:1;i:36456;b:1;i:36165;b:1;i:36508;b:1;i:36540;b:1;i:36483;b:1;i:36536;b:1;i:36543;b:1;i:36555;b:1;i:36563;b:1;i:36566;b:1;i:36409;b:1;i:36579;b:1;i:36545;b:1;i:36573;b:1;i:36588;b:1;i:36578;b:1;i:36594;b:1;i:36527;b:1;i:36600;b:1;i:36598;b:1;i:36601;b:1;i:36606;b:1;i:36608;b:1;i:36520;b:1;i:36617;b:1;i:36621;b:1;i:36510;b:1;i:36592;b:1;i:36382;b:1;i:36625;b:1;i:36662;b:1;i:36670;b:1;i:36440;b:1;i:36677;b:1;i:36449;b:1;i:36681;b:1;i:36682;b:1;i:36684;b:1;i:36611;b:1;i:36695;b:1;i:36702;b:1;i:36701;b:1;i:36703;b:1;i:36709;b:1;i:36660;b:1;i:36727;b:1;i:36718;b:1;i:36655;b:1;i:36692;b:1;i:36731;b:1;i:36708;b:1;i:36720;b:1;i:36691;b:1;i:36696;b:1;i:36639;b:1;i:36745;b:1;i:36607;b:1;i:36728;i:1;i:36741;b:1;i:36747;b:1;i:36755;b:1;i:36729;b:1;i:36757;b:1;i:36739;b:1;i:36719;b:1;i:36774;b:1;i:36599;b:1;i:36635;b:1;i:36780;b:1;i:36770;b:1;i:36782;i:1;i:36786;i:1;i:36779;b:1;i:36791;b:1;i:36793;b:1;i:36680;b:1;i:36797;b:1;i:36795;b:1;i:36798;b:1;i:36802;b:1;i:36688;b:1;i:36810;b:1;i:36811;b:1;i:36799;b:1;i:36828;b:1;i:36833;b:1;i:36806;b:1;i:36726;b:1;i:36809;b:1;i:36603;b:1;i:36845;b:1;i:36851;i:1;i:36853;b:1;i:36855;i:1;i:36872;b:1;i:36873;b:1;i:36880;b:1;i:36888;b:1;i:36896;i:1;i:36647;b:1;i:36902;b:1;i:36899;b:1;i:36551;b:1;i:36881;b:1;i:36913;b:1;i:36915;b:1;i:36914;b:1;i:36912;b:1;i:36921;b:1;i:36892;b:1;i:36860;i:1;i:36941;b:1;i:36764;b:1;i:36831;b:1;i:36945;b:1;i:36920;b:1;i:36946;b:1;i:36949;b:1;i:36951;b:1;i:36950;b:1;i:36957;b:1;i:36968;b:1;i:36965;b:1;i:36948;b:1;i:36961;i:1;i:36981;b:1;i:36980;b:1;i:36990;b:1;i:36917;b:1;i:36998;b:1;i:36993;b:1;i:36823;b:1;i:36967;b:1;i:37021;b:1;i:37024;b:1;i:36887;b:1;i:36885;b:1;i:37031;i:1;i:37009;b:1;i:36933;b:1;i:37023;b:1;i:36974;b:1;i:37050;b:1;i:37054;b:1;i:37057;b:1;i:37063;b:1;i:37046;b:1;i:37004;i:1;i:36958;b:1;i:37080;b:1;i:37093;b:1;i:37088;b:1;i:37052;b:1;i:36939;b:1;i:37097;b:1;i:36908;b:1;i:37107;b:1;i:37108;b:1;i:37106;b:1;i:37112;b:1;i:37111;b:1;i:37121;b:1;i:37125;b:1;i:37116;b:1;i:37100;b:1;i:37102;b:1;i:37138;b:1;i:37141;b:1;i:36994;b:1;i:37145;b:1;i:37067;b:1;i:37133;b:1;i:37156;b:1;i:37162;b:1;i:37164;b:1;i:37172;b:1;i:37173;b:1;i:37178;i:1;i:37151;b:1;i:37153;b:1;i:37195;b:1;i:37204;b:1;i:37163;b:1;i:37212;b:1;i:37216;b:1;i:37196;b:1;i:37203;b:1;i:37225;i:1;i:37230;b:1;i:37194;b:1;i:37118;b:1;i:37205;b:1;i:37184;b:1;i:37263;b:1;i:37265;b:1;i:37159;b:1;i:37274;b:1;i:37223;b:1;i:37286;b:1;i:37284;b:1;i:37289;b:1;i:37287;b:1;i:37165;b:1;i:37280;b:1;i:37300;b:1;i:37288;b:1;i:37239;b:1;i:37158;b:1;i:37310;b:1;i:37312;b:1;i:37309;b:1;i:37322;b:1;i:37324;b:1;i:37328;b:1;i:37327;b:1;i:37325;b:1;i:37290;b:1;i:37302;b:1;i:37349;b:1;i:37353;b:1;i:37351;b:1;i:37323;b:1;i:37357;b:1;i:37275;b:1;i:37308;b:1;i:37378;b:1;i:37387;b:1;i:37381;b:1;i:37356;b:1;i:37386;b:1;i:37182;b:1;i:37337;b:1;i:37372;b:1;i:37367;i:1;i:37417;b:1;i:37416;b:1;i:37414;b:1;i:37411;b:1;i:37412;b:1;i:37340;b:1;i:37319;b:1;i:37433;b:1;i:37441;b:1;i:37321;b:1;i:37451;b:1;i:37371;b:1;i:37446;i:1;i:37440;b:1;i:37460;b:1;i:37461;b:1;i:37403;b:1;i:37332;b:1;i:37470;b:1;i:37478;b:1;i:37477;b:1;i:37429;b:1;i:37483;b:1;i:37344;b:1;i:37485;b:1;i:37491;b:1;i:37503;b:1;i:37507;b:1;i:37404;b:1;i:37505;b:1;i:37520;b:1;i:37527;b:1;i:37528;b:1;i:37479;b:1;i:37547;b:1;i:37467;b:1;i:37529;b:1;i:37549;b:1;i:37493;i:1;i:37517;b:1;i:37564;b:1;i:37572;b:1;i:37574;b:1;i:37575;b:1;i:37567;b:1;i:37512;b:1;i:37563;b:1;i:37560;b:1;i:37559;b:1;i:37450;b:1;i:37425;b:1;i:37589;b:1;i:37595;b:1;i:37571;b:1;i:37526;b:1;i:37525;b:1;i:37600;b:1;i:37606;b:1;i:37618;i:1;i:37469;b:1;i:37621;b:1;i:37629;i:1;i:37578;b:1;i:37633;b:1;i:37496;i:1;i:37611;b:1;i:37591;b:1;i:37616;b:1;i:37612;b:1;i:37644;b:1;i:37647;b:1;i:37648;b:1;i:37458;b:1;i:37656;b:1;i:37654;b:1;i:37655;b:1;i:37569;b:1;i:37661;b:1;i:37663;b:1;i:37677;i:1;i:37673;i:1;i:37681;b:1;i:37682;b:1;i:37687;b:1;i:37532;b:1;i:37692;b:1;i:37694;b:1;i:37680;b:1;i:37702;b:1;i:37701;b:1;i:37650;b:1;i:37708;b:1;i:37651;b:1;i:37732;b:1;i:37660;b:1;i:37601;b:1;i:37734;b:1;i:37738;b:1;i:37736;b:1;i:37735;b:1;i:37620;b:1;i:37756;b:1;i:37759;b:1;i:37690;b:1;i:37766;b:1;i:37769;b:1;i:37740;b:1;i:37513;b:1;i:37704;b:1;i:37777;b:1;i:37784;b:1;i:37785;b:1;i:37741;b:1;i:37800;b:1;i:37802;b:1;i:37812;b:1;i:37761;b:1;i:37764;b:1;i:37750;b:1;i:37749;b:1;i:37821;b:1;i:37822;b:1;i:37823;b:1;i:37824;b:1;i:37831;b:1;i:37790;b:1;i:37847;b:1;i:37809;b:1;i:37852;b:1;i:37849;b:1;i:37859;b:1;i:37845;b:1;i:37870;b:1;i:37582;b:1;i:37801;i:1;i:37803;i:1;i:37811;i:1;i:37820;i:1;i:37689;b:1;i:37872;b:1;i:37892;b:1;i:37905;b:1;i:37902;b:1;i:37798;b:1;i:37883;b:1;i:37910;b:1;i:37815;b:1;i:37897;b:1;i:37919;b:1;i:37776;b:1;i:37893;b:1;i:37890;b:1;i:37935;b:1;i:37937;b:1;i:37939;b:1;i:37933;b:1;i:37969;b:1;i:37934;b:1;i:37970;b:1;i:37971;b:1;i:37973;b:1;i:37975;b:1;i:37972;b:1;i:37817;b:1;i:37860;b:1;i:37980;b:1;i:37830;b:1;i:37880;b:1;i:37985;b:1;i:37987;b:1;i:37964;b:1;i:37996;b:1;i:37989;b:1;i:37918;b:1;i:38001;b:1;i:37523;b:1;i:38004;b:1;i:37951;b:1;i:38017;i:1;i:37960;b:1;i:38002;b:1;i:37943;b:1;i:37999;b:1;i:38035;b:1;i:38008;b:1;i:38047;b:1;i:38051;b:1;i:37561;b:1;i:38011;b:1;i:38029;b:1;i:38065;b:1;i:38036;b:1;i:37898;b:1;i:38068;b:1;i:38031;b:1;i:38072;b:1;i:37993;b:1;i:38069;b:1;i:38026;b:1;i:38034;b:1;i:38059;b:1;i:38101;b:1;i:38112;b:1;i:38114;b:1;i:38042;b:1;i:38073;b:1;i:37990;b:1;i:38130;b:1;i:38143;b:1;i:38092;b:1;i:37948;b:1;i:38136;b:1;i:38150;b:1;i:38148;b:1;i:38153;b:1;i:38071;b:1;i:38125;b:1;i:38127;b:1;i:38056;b:1;i:38158;b:1;i:38160;b:1;i:38138;b:1;i:38169;b:1;i:38171;b:1;i:38174;b:1;i:38186;b:1;i:38151;b:1;i:38163;b:1;i:38193;b:1;i:38192;b:1;i:38062;b:1;i:38111;b:1;i:38159;b:1;i:38212;b:1;i:38183;b:1;i:38208;b:1;i:38203;b:1;i:38230;b:1;i:38223;b:1;i:38236;b:1;i:38227;b:1;i:38237;b:1;i:38225;b:1;i:38238;b:1;i:38235;b:1;i:38247;b:1;i:38253;b:1;i:38251;b:1;i:38210;b:1;i:38197;b:1;i:38216;b:1;i:38232;b:1;i:38233;b:1;i:38268;b:1;i:38271;b:1;i:38269;b:1;i:38273;b:1;i:38276;b:1;i:38272;b:1;i:38281;b:1;i:38287;b:1;i:38152;b:1;i:38285;b:1;i:38289;b:1;i:38302;b:1;i:38301;b:1;i:38102;b:1;i:38307;b:1;i:38310;b:1;i:38214;b:1;i:38314;b:1;i:38304;b:1;i:38316;b:1;i:38315;b:1;i:38323;b:1;i:38328;b:1;i:38189;b:1;i:38283;b:1;i:38334;b:1;i:38280;b:1;i:38336;b:1;i:38245;b:1;i:38345;b:1;i:38179;b:1;i:38205;b:1;i:38350;b:1;i:38351;b:1;i:38330;i:1;i:38367;b:1;i:38374;b:1;i:38378;b:1;i:38373;b:1;i:38369;b:1;i:38386;b:1;i:38275;b:1;i:38348;b:1;i:38399;b:1;i:38400;b:1;i:38405;b:1;i:38392;b:1;i:38401;b:1;i:38402;b:1;i:38410;i:1;i:38368;b:1;i:38420;b:1;i:38417;b:1;i:38422;b:1;i:38390;b:1;i:38431;i:1;i:38442;b:1;i:38476;b:1;i:38471;b:1;i:38187;b:1;i:38499;b:1;i:38504;b:1;i:38497;b:1;i:38505;b:1;i:38509;b:1;i:38495;b:1;i:38408;i:1;i:38522;i:1;i:38507;b:1;i:38502;b:1;i:38461;b:1;i:38530;b:1;i:38531;b:1;i:38514;b:1;i:38512;b:1;i:38465;b:1;i:38539;i:1;i:38542;b:1;i:38537;b:1;i:38559;b:1;i:38560;b:1;i:38535;b:1;i:38459;b:1;i:38520;b:1;i:38479;b:1;i:38552;b:1;i:38587;b:1;i:38593;b:1;i:38596;b:1;i:38571;b:1;i:38599;b:1;i:38601;b:1;i:38576;i:1;i:38579;i:1;i:38517;b:1;i:38605;b:1;i:38611;b:1;i:38586;b:1;i:38563;b:1;i:38618;b:1;i:38510;b:1;i:38607;i:1;i:38485;b:1;i:38644;b:1;i:38352;b:1;i:38591;b:1;i:38660;b:1;i:38649;b:1;i:38647;b:1;i:38661;b:1;i:38671;b:1;i:38657;b:1;i:38679;b:1;i:38684;b:1;i:38682;b:1;i:38604;i:1;i:38694;b:1;i:38696;b:1;i:38669;b:1;i:38674;b:1;i:38701;b:1;i:38703;b:1;i:38630;i:1;i:38688;b:1;i:38719;b:1;i:38713;b:1;i:38720;b:1;i:38724;b:1;i:38487;b:1;i:38656;b:1;i:38702;b:1;i:38740;b:1;i:38738;b:1;i:38769;b:1;i:1602;b:1;i:38777;b:1;i:38780;b:1;i:38758;b:1;i:38756;b:1;i:38782;b:1;i:38790;b:1;i:38800;b:1;i:38798;b:1;i:38792;i:1;i:38811;b:1;i:38795;b:1;i:38799;b:1;i:38784;b:1;i:38849;b:1;i:38859;i:1;i:38862;b:1;i:38878;b:1;i:38886;b:1;i:38880;b:1;i:38898;b:1;i:38897;b:1;i:38817;b:1;i:38907;b:1;i:38906;b:1;i:38845;b:1;i:38913;b:1;i:38910;b:1;i:38924;b:1;i:38923;b:1;i:38927;b:1;i:38930;b:1;i:38929;b:1;i:38933;b:1;i:38931;b:1;i:38920;b:1;i:38909;b:1;i:38890;b:1;i:38964;b:1;i:38973;b:1;i:38975;b:1;i:38984;b:1;i:38987;b:1;i:38971;b:1;i:38946;b:1;i:38991;b:1;i:38990;b:1;i:38994;b:1;i:38980;b:1;i:38883;b:1;i:39018;b:1;i:39005;b:1;i:39003;b:1;i:39021;b:1;i:39019;b:1;i:39030;b:1;i:39041;b:1;i:38950;b:1;i:38962;b:1;i:38957;b:1;i:39040;b:1;i:39046;b:1;i:39049;b:1;i:39053;b:1;i:38943;b:1;i:39059;b:1;i:39060;b:1;i:39063;b:1;i:39062;b:1;i:39032;b:1;i:38853;b:1;i:39081;b:1;i:39084;b:1;i:38802;b:1;i:38870;b:1;i:38917;b:1;i:39106;b:1;i:39100;b:1;i:39110;b:1;i:38708;b:1;i:39117;b:1;i:39045;b:1;i:39120;b:1;i:39065;b:1;i:39073;b:1;i:39135;b:1;i:39136;b:1;i:39137;b:1;i:39146;b:1;i:39080;b:1;i:39151;b:1;i:39150;b:1;i:39016;b:1;i:38989;b:1;i:39112;b:1;i:39113;b:1;i:39172;b:1;i:39189;b:1;i:39194;b:1;i:39220;b:1;i:39231;b:1;i:39107;b:1;i:39209;b:1;i:39244;b:1;i:39228;b:1;i:39240;b:1;i:39138;b:1;i:39267;b:1;i:39272;b:1;i:39273;b:1;i:39274;i:1;i:39247;b:1;i:39254;b:1;i:39294;b:1;i:39291;b:1;i:39292;b:1;i:39300;b:1;i:39207;b:1;i:39125;b:1;i:39320;b:1;i:39187;i:1;i:39278;i:1;i:39325;b:1;i:39328;b:1;i:39349;b:1;i:39352;b:1;i:39350;b:1;i:39360;b:1;i:39077;b:1;i:39298;b:1;i:39361;b:1;i:39368;b:1;i:39348;b:1;i:39372;b:1;i:39293;b:1;i:39380;b:1;i:39375;b:1;i:39385;b:1;i:39261;b:1;i:39393;b:1;i:39395;b:1;i:39398;b:1;i:39392;b:1;i:39337;b:1;i:39334;b:1;i:39414;b:1;i:39271;b:1;i:39420;b:1;i:39435;b:1;i:39425;b:1;i:39441;b:1;i:39449;b:1;i:39448;i:1;i:39452;b:1;i:39455;b:1;i:39438;b:1;i:39454;b:1;i:39426;b:1;i:39433;b:1;i:39440;b:1;i:39468;b:1;i:39469;b:1;i:39472;b:1;i:39477;b:1;i:39384;b:1;i:39504;b:1;i:39506;b:1;i:39475;b:1;i:39485;b:1;i:39523;b:1;i:39371;b:1;i:39536;b:1;i:39552;b:1;i:39549;b:1;i:39559;b:1;i:39557;b:1;i:39359;b:1;i:39566;b:1;i:39554;b:1;i:39531;b:1;i:39570;b:1;i:39607;b:1;i:39605;b:1;i:39601;b:1;i:39612;i:1;i:39502;b:1;i:39558;b:1;i:39651;b:1;i:39652;b:1;i:39629;b:1;i:39551;b:1;i:39656;b:1;i:39657;b:1;i:39567;b:1;i:39662;b:1;i:39672;b:1;i:39678;b:1;i:39680;b:1;i:39537;b:1;i:39700;b:1;i:39683;b:1;i:39556;b:1;i:39617;b:1;i:39716;b:1;i:39696;b:1;i:39519;b:1;i:39731;b:1;i:39670;b:1;i:39740;b:1;i:39745;b:1;i:39722;b:1;i:39747;b:1;i:39596;b:1;i:39533;b:1;i:39738;b:1;i:39708;b:1;i:39711;b:1;i:39765;b:1;i:39766;b:1;i:39773;b:1;i:39772;b:1;i:39787;b:1;i:39715;b:1;i:39791;b:1;i:39667;b:1;i:39801;b:1;i:39800;b:1;i:39802;b:1;i:39770;b:1;i:39807;b:1;i:39809;b:1;i:39365;b:1;i:39592;b:1;i:39767;b:1;i:39790;b:1;i:39841;b:1;i:39844;b:1;i:39799;b:1;i:39842;b:1;i:39851;b:1;i:39856;b:1;i:39857;b:1;i:39862;b:1;i:39863;b:1;i:39866;b:1;i:39871;b:1;i:39874;b:1;i:39881;b:1;i:39885;i:1;i:39887;i:1;i:39892;b:1;i:39898;b:1;i:39872;b:1;i:39865;b:1;i:39756;b:1;i:39820;b:1;i:39915;b:1;i:39920;b:1;i:39777;b:1;i:39940;b:1;i:39855;b:1;i:39782;b:1;i:39951;b:1;i:39925;b:1;i:39923;b:1;i:39900;b:1;i:39967;b:1;i:39969;b:0;i:39918;b:1;i:39937;i:1;i:39965;b:1;i:39992;b:1;i:39997;b:1;i:39995;b:1;i:39994;b:1;i:39701;b:1;i:40014;b:1;i:40018;b:1;i:40029;b:1;i:40021;b:1;i:40050;b:1;i:40053;b:1;i:39861;b:1;i:40044;b:1;i:40068;b:1;i:40070;b:1;i:40065;b:1;i:40087;b:1;i:39996;b:1;i:40088;b:1;i:39956;b:1;i:39905;b:1;i:39908;b:1;i:40000;b:1;i:40103;b:1;i:40104;i:1;i:40124;b:1;i:40049;b:1;i:40144;b:1;i:40129;b:1;i:40185;b:1;i:39784;b:1;i:39639;b:1;i:40165;b:1;i:40175;b:1;i:40177;b:1;i:40178;b:1;i:40209;b:1;i:40211;b:1;i:40219;b:1;i:40221;b:1;i:40229;b:1;i:40218;b:1;i:40181;b:1;i:40253;b:1;i:40256;b:1;i:40233;b:1;i:40263;b:1;i:40223;b:1;i:40265;b:1;i:40269;b:1;i:40274;b:1;i:40197;b:1;i:40293;b:1;i:40202;b:1;i:40300;b:1;i:40237;b:1;i:40299;b:1;i:40320;b:1;i:40336;b:1;i:40337;b:1;i:40340;b:1;i:40343;b:1;i:40359;b:1;i:40364;b:1;i:40356;b:1;i:40261;b:1;i:40370;b:1;i:40373;b:1;i:40377;b:1;i:40380;b:1;i:40367;b:1;i:40400;b:1;i:40395;i:1;i:40422;b:1;i:40426;b:1;i:40270;b:1;i:40243;b:1;i:40440;i:1;i:40417;b:1;i:40454;b:1;i:40461;b:1;i:40459;b:1;i:40467;b:1;i:40474;b:1;i:40283;b:1;i:40396;b:1;i:40495;b:1;i:40494;b:1;i:40490;b:1;i:40469;b:1;i:40171;i:1;i:40517;b:1;i:40473;b:1;i:40372;b:1;i:40534;b:1;i:40538;b:1;i:40543;b:1;i:40540;b:1;i:40346;b:1;i:40485;i:1;i:40451;b:1;i:40564;b:1;i:40463;b:1;i:40325;b:1;i:40550;b:1;i:40569;b:1;i:40572;b:1;i:40574;b:1;i:40587;b:1;i:40607;b:1;i:40613;b:1;i:40588;b:1;i:40508;i:1;i:40514;i:1;i:40633;b:1;i:40634;b:1;i:40644;b:1;i:40393;b:1;i:40657;b:1;i:40670;b:1;i:40669;b:1;i:40672;b:1;i:40676;b:1;i:40636;i:1;i:40578;b:1;i:40579;b:1;i:40516;b:1;i:40668;i:1;i:40694;b:1;i:40628;b:1;i:40701;b:1;i:40606;b:1;i:40711;b:1;i:40665;b:1;i:40427;b:1;i:40547;b:1;i:40563;b:1;i:40726;b:1;i:40640;i:1;i:40732;i:1;i:40746;b:1;i:40748;b:1;i:40671;b:1;i:40750;b:1;i:40749;b:1;i:40751;b:1;i:40632;b:1;i:40756;b:1;i:40754;b:1;i:40757;b:1;i:40752;b:1;i:40675;b:1;i:40724;b:1;i:40709;b:1;i:40794;b:1;i:40807;b:1;i:40809;b:1;i:40795;b:1;i:40806;b:1;i:40810;b:1;i:40817;b:1;i:40812;b:1;i:40820;b:1;i:40825;b:1;i:40766;b:1;i:40801;i:1;i:40854;b:1;i:40661;b:1;i:40865;b:1;i:40866;b:1;i:40864;b:1;i:40882;b:1;i:40881;b:1;i:40730;i:1;i:40656;b:1;i:40893;b:1;i:40876;i:1;i:40904;b:1;i:40907;b:1;i:40910;b:1;i:40914;b:1;i:40915;b:1;i:40933;b:1;i:40935;b:1;i:40762;b:1;i:40936;b:1;i:40957;b:1;i:40901;i:1;i:40873;i:1;i:40943;b:1;i:40952;b:1;i:40965;b:1;i:40970;b:1;i:40975;b:1;i:40942;b:1;i:40978;b:1;i:40949;b:1;i:40887;b:1;i:40985;b:1;i:40894;b:1;i:40998;b:1;i:41012;b:1;i:41019;b:1;i:41015;b:1;i:41028;b:1;i:41017;b:1;i:41037;b:1;i:41038;b:1;i:41023;b:1;i:41052;b:1;i:41011;b:1;i:40920;b:1;i:41063;i:1;i:41046;b:1;i:41075;b:1;i:41058;b:1;i:41080;b:1;i:41056;b:1;i:41088;b:1;i:41097;b:1;i:41098;b:1;i:41009;b:1;i:41100;b:1;i:41102;b:1;i:41104;b:1;i:40954;b:1;i:40991;b:1;i:41114;b:1;i:41106;i:1;i:41072;b:1;i:41068;b:1;i:41002;b:1;i:41129;b:1;i:41093;b:1;i:41103;b:1;i:41140;b:1;i:41143;i:1;i:40971;b:1;i:41145;b:1;i:41148;b:1;i:41167;b:1;i:41166;b:1;i:41134;b:1;i:41170;b:1;i:41175;b:1;i:41190;b:1;i:41193;b:1;i:41187;b:1;i:41201;b:1;i:41202;b:1;i:41094;b:1;i:41204;b:1;i:41177;b:1;i:41078;b:1;i:41221;b:1;i:41227;b:1;i:41232;b:1;i:41243;b:1;i:41235;b:1;i:41213;b:1;i:41242;b:1;i:41250;b:1;i:41253;b:1;i:41194;i:1;i:41197;i:1;i:41228;b:1;i:41021;b:1;i:41081;b:1;i:41211;b:1;i:41278;b:1;i:41280;b:1;i:41285;b:1;i:41288;b:1;i:41289;b:1;i:41218;b:1;i:41251;b:1;i:41293;b:1;i:41199;b:1;i:41308;b:1;i:41312;b:1;i:41269;b:1;i:41282;b:1;i:41321;b:1;i:41325;i:1;i:41117;b:1;i:41330;b:1;i:41343;b:1;i:41366;i:1;i:41390;i:1;i:41352;b:1;i:41151;b:1;i:41073;b:1;i:41158;b:1;i:41395;i:1;i:41435;b:1;i:41439;i:1;i:41489;b:1;i:41277;b:1;i:41488;i:1;i:41299;b:1;i:41302;b:1;i:41358;b:1;i:41521;b:1;i:41533;b:1;i:41516;b:1;i:41499;b:1;i:41421;b:1;i:41478;i:1;i:41192;b:1;i:41564;b:1;i:41567;b:1;i:41573;b:1;i:41556;b:1;i:41577;b:1;i:41580;b:1;i:41596;b:1;i:41506;b:1;i:41597;b:1;i:41602;b:1;i:41614;b:1;i:41623;b:1;i:41632;b:1;i:41637;b:1;i:41309;b:1;i:41643;b:1;i:41529;b:1;i:41635;b:1;i:41697;b:1;i:41616;b:1;i:41663;b:1;i:41686;b:1;i:41718;b:1;i:41719;b:1;i:41570;i:1;i:41715;b:1;i:27278;b:1;i:41571;b:1;i:41741;b:1;i:41738;b:1;i:41745;b:1;i:41752;b:1;i:41648;b:1;i:41754;b:1;i:41654;b:1;i:41762;b:1;i:41582;i:1;i:41776;b:1;i:41783;b:1;i:41792;b:1;i:41791;b:1;i:41785;b:1;i:41683;b:1;i:41786;b:1;i:41801;b:1;i:41806;b:1;i:41827;b:1;i:41780;i:1;i:41844;b:1;i:41843;b:1;i:41851;b:1;i:41828;b:1;i:41840;b:1;i:41604;b:1;i:41890;b:1;i:41892;b:1;i:41561;b:1;i:41725;b:1;i:41908;b:1;i:41930;b:1;i:41895;i:1;i:41838;i:1;i:41932;b:1;i:41954;b:1;i:41966;b:1;i:41967;b:1;i:41979;b:1;i:41750;b:1;i:41645;b:1;i:41789;b:1;i:41797;b:1;i:41994;b:1;i:41990;b:1;i:41996;b:1;i:41970;b:1;i:42003;b:1;i:41950;b:1;i:42016;b:1;i:42009;b:1;i:42017;b:1;i:41998;i:1;i:42035;b:1;i:42033;b:1;i:41964;b:1;i:42045;b:1;i:41810;b:1;i:42000;b:1;i:42061;b:1;i:42066;b:1;i:42068;b:1;i:42067;b:1;i:41808;b:1;i:41753;b:1;i:42018;b:1;i:42076;b:1;i:42086;b:1;i:42081;b:1;i:42092;b:1;i:42098;b:1;i:41938;b:1;i:42110;b:1;i:42112;b:1;i:42115;b:1;i:42134;b:1;i:42136;b:1;i:42138;b:1;i:42152;b:1;i:42150;b:1;i:42151;b:1;i:42153;b:1;i:42155;b:1;i:42100;b:1;i:42102;b:1;i:42157;b:1;i:42122;i:1;i:42147;b:1;i:41769;b:1;i:41777;b:1;i:41952;b:1;i:42173;b:1;i:42154;b:1;i:42183;b:1;i:42186;b:1;i:42047;b:1;i:42188;b:1;i:41730;b:1;i:42221;b:1;i:42224;b:1;i:42227;b:1;i:42187;b:1;i:42226;b:1;i:42223;b:1;i:42230;b:1;i:42127;i:1;i:42208;i:1;i:42205;i:1;i:42251;b:1;i:42234;b:1;i:42263;b:1;i:42262;b:1;i:42172;b:1;i:42228;b:1;i:42273;b:1;i:42271;b:1;i:42196;b:1;i:42229;b:1;i:42106;b:1;i:42286;b:1;i:42285;b:1;i:41713;b:1;i:42296;b:1;i:42239;b:1;i:42160;i:1;i:42267;b:1;i:42181;b:1;i:42144;b:1;i:42323;b:1;i:42284;b:1;i:42291;i:1;i:42299;i:1;i:42335;b:1;i:42325;b:1;i:42324;b:1;i:42347;b:1;i:42328;b:1;i:42354;b:1;i:42320;b:1;i:42326;b:1;i:42339;b:1;i:42165;b:1;i:42368;b:1;i:42303;b:1;i:42372;b:1;i:42375;b:1;i:42378;b:1;i:42380;b:1;i:42390;i:1;i:42391;i:1;i:42384;i:1;i:42407;b:1;i:42364;b:1;i:42416;b:1;i:42439;b:1;i:42049;b:1;i:42237;b:1;i:42015;b:1;i:42444;b:1;i:42359;b:1;i:42111;b:1;i:42385;b:1;i:42454;b:1;i:42455;b:1;i:42458;b:1;i:42461;b:1;i:42457;b:1;i:42410;b:1;i:42484;b:1;i:42498;b:1;i:42503;b:1;i:42449;b:1;i:42505;b:1;i:42504;b:1;i:42398;b:1;i:13777;b:1;i:42519;b:1;i:42477;i:1;i:42525;b:1;i:42453;b:1;i:42532;b:1;i:42540;b:1;i:42543;b:1;i:42550;b:1;i:42554;b:1;i:42557;b:1;i:42558;b:1;i:42560;b:1;i:42447;b:1;i:42522;b:1;i:42567;b:1;i:42530;b:1;i:42568;b:1;i:42555;i:1;i:42538;b:1;i:42585;b:1;i:42582;b:1;i:42589;b:1;i:42590;b:1;i:42469;i:1;i:42474;i:1;i:42600;b:1;i:42602;b:1;i:42601;b:1;i:42593;i:1;i:42609;b:1;i:42612;b:1;i:42614;b:1;i:42566;b:1;i:42586;b:1;i:42408;b:1;i:42459;b:1;i:42591;b:1;i:42638;b:1;i:42643;i:1;i:42640;b:1;i:42517;i:1;i:42655;b:1;i:42662;b:1;i:42669;b:1;i:42687;b:1;i:42657;b:1;i:42685;b:1;i:42659;b:1;i:42648;i:1;i:42645;i:1;i:42718;b:1;i:42722;b:1;i:42633;b:1;i:42275;b:1;i:42730;b:1;i:42741;b:1;i:42740;b:1;i:42752;b:1;i:42753;i:1;i:42665;b:1;i:42507;b:1;i:42723;b:1;i:42769;b:1;i:42725;b:1;i:42775;b:1;i:42721;b:1;i:42404;b:1;i:42788;b:1;i:42793;b:1;i:42735;b:1;i:42787;b:1;i:42821;b:1;i:42833;b:1;i:42830;b:1;i:42699;i:1;i:42750;i:1;i:42840;i:1;i:42836;b:1;i:42805;b:1;i:42852;b:1;i:42858;b:1;i:42868;b:1;i:42871;b:1;i:42874;b:1;i:42875;b:1;i:42826;b:1;i:42877;b:1;i:42880;b:1;i:42598;b:1;i:42781;b:1;i:42599;b:1;i:42905;b:1;i:42907;b:1;i:42915;b:1;i:42910;b:1;i:42916;b:1;i:42814;b:1;i:42925;b:1;i:42893;b:1;i:42802;b:1;i:42931;b:1;i:42572;b:1;i:42955;b:1;i:42789;b:1;i:42960;b:1;i:42628;b:1;i:42950;b:1;i:42977;b:1;i:42999;b:1;i:42854;b:1;i:43009;b:1;i:42968;b:1;i:42967;b:1;i:42864;b:1;i:43023;b:1;i:43042;b:1;i:42674;b:1;i:42818;b:1;i:43026;b:1;i:42597;b:1;i:43055;b:1;i:43056;b:1;i:42923;b:1;i:42908;b:1;i:43079;b:1;i:43078;b:1;i:43069;b:1;i:43080;b:1;i:43105;b:1;i:43128;b:1;i:43131;b:1;i:43109;i:1;i:43050;b:1;i:43123;b:1;i:43146;b:1;i:43121;b:1;i:43149;b:1;i:42911;b:1;i:43156;b:1;i:43161;b:1;i:42717;b:1;i:43168;b:1;i:43143;b:1;i:43024;b:1;i:43175;b:1;i:43018;b:1;i:43081;i:1;i:43102;i:1;i:43147;b:1;i:43198;b:1;i:43199;b:1;i:42887;b:1;i:43179;i:1;i:43214;i:1;i:43133;b:1;i:43217;b:1;i:43165;b:1;i:43162;b:1;i:43115;b:1;i:43119;b:1;i:43159;b:1;i:42810;b:1;i:43166;b:1;i:43208;b:1;i:43041;b:1;i:43038;b:1;i:43151;b:1;i:43240;b:1;i:43141;b:1;i:43266;b:1;i:43275;b:1;i:43299;b:1;i:43311;b:1;i:43318;b:1;i:43294;b:1;i:43323;b:1;i:43302;b:1;i:29535;i:1;i:43329;i:1;i:38635;i:1;i:29488;i:1;i:43022;b:1;i:43334;b:1;i:42914;b:1;i:43344;b:1;i:43127;b:1;i:43355;i:1;i:43031;b:1;i:43362;b:1;i:43376;b:1;i:43254;b:1;i:43253;b:1;i:43384;b:1;i:43383;b:1;i:43387;b:1;i:43382;b:1;i:42758;i:1;i:43243;b:1;i:43281;b:1;i:43399;b:1;i:43412;b:1;i:43421;b:1;i:43424;b:1;i:43068;b:1;i:43283;b:1;i:43258;b:1;i:43447;b:1;i:43452;b:1;i:43455;b:1;i:43458;b:1;i:43358;i:1;i:43470;b:1;i:43469;b:1;i:43464;b:1;i:43473;b:1;i:43476;b:1;i:43285;b:1;i:43287;b:1;i:43407;b:1;i:43489;b:1;i:43502;b:1;i:43504;b:1;i:43377;b:1;i:43379;b:1;i:43509;b:1;i:43510;b:1;i:43519;b:1;i:43426;b:1;i:43528;b:1;i:43541;b:1;i:43547;b:1;i:43370;b:1;i:43465;b:1;i:43570;b:1;i:43515;b:1;i:43512;b:1;i:43585;b:1;i:43565;b:1;i:43536;b:1;i:43564;b:1;i:43600;b:1;i:43599;b:1;i:43602;b:1;i:43601;b:1;i:43598;b:1;i:43607;b:1;i:43604;b:1;i:43606;b:1;i:43605;b:1;i:43588;b:1;i:43613;b:1;i:43582;b:1;i:43624;b:1;i:43628;b:1;i:43638;b:1;i:43516;b:1;i:43649;b:1;i:43654;b:1;i:43645;b:1;i:43658;b:1;i:43660;b:1;i:43675;b:1;i:43673;b:1;i:43615;b:1;i:43685;b:1;i:43577;b:1;i:43688;b:1;i:43690;b:1;i:43692;b:1;i:43700;b:1;i:43699;b:1;i:43721;b:1;i:43431;b:1;i:43579;b:1;i:43756;b:1;i:43757;b:1;i:43524;b:1;i:43754;b:1;i:43770;b:1;i:43693;b:1;i:43768;b:1;i:43581;i:1;i:43753;b:1;i:43785;b:1;i:43548;b:1;i:43786;b:1;i:43787;b:1;i:43571;b:1;i:43817;b:1;i:43780;i:1;i:43816;b:1;i:43818;i:1;i:43722;b:1;i:43823;b:1;i:43810;b:1;i:43852;b:1;i:43862;b:1;i:43837;b:1;i:43631;b:1;i:43812;b:1;i:43885;i:1;i:43853;b:1;i:43798;b:1;i:43912;b:1;i:43864;b:1;i:43763;b:1;i:43943;b:1;i:43845;b:1;i:43945;b:1;i:43958;b:1;i:43735;b:1;i:43687;b:1;i:43962;b:1;i:43904;b:1;i:43948;i:1;i:43972;b:1;i:43961;b:1;i:43954;i:1;i:43977;b:1;i:43936;b:1;i:43964;b:1;i:43999;b:1;i:43997;b:1;i:44004;b:1;i:43986;b:1;i:44019;b:1;i:44002;b:1;i:44027;b:1;i:43856;b:1;i:44028;b:1;i:44036;b:1;i:23569;i:1;i:44046;b:1;i:44049;b:1;i:43995;b:1;i:43990;b:1;i:44033;b:1;i:43981;b:1;i:43942;b:1;i:43908;b:1;i:44072;b:1;i:44078;b:1;i:44083;b:1;i:44082;b:1;i:44094;b:1;i:44091;i:1;i:44107;b:1;i:44113;b:1;i:44042;b:1;i:43939;b:1;i:44142;b:1;i:44119;b:1;i:44061;b:1;i:44163;b:1;i:44152;b:1;i:44159;b:1;i:43970;i:1;i:44174;b:1;i:44063;b:1;i:44182;i:1;i:44189;b:1;i:44054;b:1;i:44200;b:1;i:44181;b:1;i:44203;b:1;i:44217;b:1;i:44216;b:1;i:44218;b:1;i:44228;b:1;i:44236;b:1;i:44241;b:1;i:44104;i:1;i:44272;b:1;i:44271;b:1;i:44273;b:1;i:44275;b:1;i:44279;b:1;i:44232;b:1;i:44288;b:1;i:44247;b:1;i:44240;b:1;i:44303;b:1;i:44306;b:1;i:44321;b:1;i:43684;b:1;i:44188;b:1;i:44322;b:1;i:44323;b:1;i:44305;b:1;i:44206;b:1;i:44199;b:1;i:44302;b:1;i:44340;b:1;i:44344;b:1;i:44331;b:1;i:44339;b:1;i:44295;b:1;i:44389;b:1;i:44392;b:1;i:44393;b:1;i:44400;i:1;i:44419;b:1;i:44422;b:1;i:44417;b:1;i:44193;b:1;i:44192;b:1;i:44387;b:1;i:44354;b:1;i:44434;b:1;i:44368;b:1;i:44320;b:1;i:44327;b:1;i:44413;b:1;i:44455;b:1;i:44453;b:1;i:44157;b:1;i:44355;b:1;i:44391;b:1;i:41436;i:1;i:44375;b:1;i:44481;b:1;i:44299;b:1;i:44298;b:1;i:44041;b:1;i:44238;b:1;i:44516;b:1;i:44209;b:1;i:44492;b:1;i:44491;b:1;i:44522;b:1;i:44536;b:1;i:44505;b:1;i:44553;b:1;i:44555;b:1;i:44507;b:1;i:44581;b:1;i:44586;b:1;i:44591;b:1;i:44598;b:1;i:44589;b:1;i:44624;b:1;i:44625;b:1;i:44378;b:1;i:44353;b:1;i:44590;b:1;i:44612;b:1;i:44450;b:1;i:44669;b:1;i:44673;b:1;i:44672;b:1;i:44677;b:1;i:44682;b:1;i:44683;b:1;i:44684;b:1;i:44685;b:1;i:44693;b:1;i:44694;b:1;i:44695;b:1;i:44471;b:1;i:44477;b:1;i:44702;b:1;i:44704;b:1;i:44708;b:1;i:44488;b:1;i:44715;b:1;i:44726;b:1;i:44732;b:1;i:44733;b:1;i:44736;b:1;i:44737;b:1;i:44346;b:1;i:44744;b:1;i:44745;b:1;i:44720;b:1;i:44696;b:1;i:44755;b:1;i:44723;b:1;i:44759;b:1;i:44764;b:1;i:44767;b:1;i:44768;b:1;i:44675;b:1;i:44780;b:1;i:44582;b:1;i:44765;b:1;i:44689;b:1;i:44758;b:1;i:44762;b:1;i:44642;b:1;i:44776;b:1;i:44799;b:1;i:44811;b:1;i:44819;b:1;i:44825;b:1;i:44827;b:1;i:44833;b:1;i:44836;b:1;i:44619;b:1;i:44839;b:1;i:44771;i:1;i:44703;b:1;i:44823;b:1;i:44851;b:1;i:44197;b:1;i:44637;b:1;i:44857;b:1;i:44446;b:1;i:44458;b:1;i:44863;b:1;i:44866;b:1;i:44831;b:1;i:44869;b:1;i:44868;b:1;i:44871;b:1;i:44797;b:1;i:44882;b:1;i:44883;b:1;i:44753;b:1;i:44639;b:1;i:44743;b:1;i:44897;b:1;i:44899;b:1;i:44875;b:1;i:44909;b:1;i:44910;b:1;i:44877;b:1;i:44923;b:1;i:44801;b:1;i:44928;b:1;i:44933;b:1;i:44930;b:1;i:44934;b:1;i:44932;b:1;i:44939;b:1;i:44865;b:1;i:44943;b:1;i:44946;b:1;i:44948;b:1;i:44958;b:1;i:44960;b:1;i:44891;b:1;i:44961;b:1;i:44791;b:1;i:44964;b:1;i:44970;b:1;i:44900;b:1;i:44991;b:1;i:44951;b:1;i:44976;b:1;i:44996;b:1;i:44870;b:1;i:43933;b:1;i:45010;b:1;i:45007;b:1;i:45015;b:1;i:45021;b:1;i:45018;b:1;i:45026;b:1;i:45037;b:1;i:45040;b:1;i:45049;b:1;i:45056;b:1;i:45057;b:1;i:45071;b:1;i:45075;b:1;i:45083;b:1;i:45089;b:1;i:45092;b:1;i:45096;b:1;i:45097;b:1;i:45101;b:1;i:45098;b:1;i:45088;b:1;i:44901;b:1;i:45032;b:1;i:45036;b:1;i:45070;b:1;i:45126;b:1;i:45129;b:1;i:45117;b:1;i:45141;b:1;i:45080;b:1;i:45146;b:1;i:45039;b:1;i:45106;b:1;i:45163;b:1;i:45133;b:1;i:45171;b:1;i:45170;b:1;i:44971;b:1;i:44999;b:1;i:45160;b:1;i:45184;b:1;i:45185;b:1;i:45192;b:1;i:45207;b:1;i:45205;b:1;i:45201;b:1;i:45215;b:1;i:45186;b:1;i:45222;b:1;i:45182;b:1;i:45226;b:1;i:45242;b:1;i:45239;b:1;i:45251;b:1;i:45259;b:1;i:45102;b:1;i:45270;b:1;i:45249;b:1;i:45277;b:1;i:45279;b:1;i:45280;b:1;i:45230;b:1;i:45214;b:1;i:45284;b:1;i:45072;b:1;i:45293;b:1;i:45166;b:1;i:45167;b:1;i:45295;b:1;i:45297;b:1;i:45296;b:1;i:45278;b:1;i:45304;b:1;i:45303;b:1;i:45292;b:1;i:45125;b:1;i:45294;b:1;i:45329;b:1;i:45345;b:1;i:45347;b:1;i:45352;b:1;i:45330;b:1;i:45337;b:1;i:45377;b:1;i:45333;b:1;i:45388;b:1;i:45390;b:1;i:45331;b:1;i:45375;b:1;i:45355;b:1;i:45366;b:1;i:45362;b:1;i:45391;b:1;i:45384;b:1;i:45415;b:1;i:45411;b:1;i:45426;b:1;i:45378;b:1;i:45177;b:1;i:45440;b:1;i:45439;b:1;i:45412;b:1;i:45444;b:1;i:45460;b:1;i:45349;b:1;i:45466;b:1;i:45470;b:1;i:45474;b:1;i:45154;b:1;i:45420;b:1;i:45476;b:1;i:45492;b:1;i:45494;b:1;i:45322;b:1;i:45496;b:1;i:45498;b:1;i:45499;b:1;i:45449;b:1;i:45481;b:1;i:45506;b:1;i:45429;b:1;i:45364;b:1;i:45516;b:1;i:45518;b:1;i:45435;b:1;i:45523;b:1;i:45546;b:1;i:45545;b:1;i:45539;b:1;i:45469;b:1;i:45560;b:1;i:45517;b:1;i:45493;b:1;i:45515;b:1;i:44805;b:1;i:45549;b:1;i:45550;b:1;i:45584;b:1;i:45585;b:1;i:45588;b:1;i:45592;b:1;i:45424;b:1;i:45543;b:1;i:45603;b:1;i:45602;b:1;i:45618;b:1;i:45632;b:1;i:45635;b:1;i:45641;b:1;i:45648;b:1;i:45649;b:1;i:45656;b:1;i:45627;b:1;i:45528;b:1;i:45620;b:1;i:45610;b:1;i:45671;b:1;i:45667;b:1;i:45653;b:1;i:45703;b:1;i:45710;b:1;i:45659;b:1;i:45698;b:1;i:45717;b:1;i:45687;b:1;i:45565;b:1;i:45647;b:1;i:45754;b:1;i:45570;b:1;i:45608;b:1;i:45607;b:1;i:45756;b:1;i:45686;b:1;i:45763;b:1;i:45682;b:1;i:45768;b:1;i:45777;b:1;i:45773;b:1;i:45638;b:1;i:45800;b:1;i:45804;b:1;i:45803;b:1;i:45797;b:1;i:45801;b:1;i:45810;b:1;i:45809;b:1;i:45814;b:1;i:45668;b:1;i:45833;b:1;i:45631;b:1;i:45834;b:1;i:45845;b:1;i:45853;b:1;i:45849;b:1;i:45808;b:1;i:45859;b:1;i:45863;b:1;i:45865;i:1;i:45871;b:1;i:45793;b:1;i:45757;b:1;i:45884;b:1;i:45899;b:1;i:45765;b:1;i:45877;b:1;i:45913;b:1;i:45919;b:1;i:45874;b:1;i:45926;b:1;i:45921;b:1;i:45933;b:1;i:45937;b:1;i:45860;b:1;i:45612;b:1;i:45947;b:1;i:45966;b:1;i:45731;b:1;i:45901;b:1;i:45997;b:1;i:45995;b:1;i:46002;i:1;i:46013;b:1;i:45939;b:1;i:45609;b:1;i:46027;b:1;i:46026;b:1;i:46042;b:1;i:45858;b:1;i:46039;b:1;i:45959;b:1;i:46085;b:1;i:46089;b:1;i:46037;b:1;i:46106;b:1;i:46107;b:1;i:46101;b:1;i:46108;b:1;i:46109;b:1;i:46061;b:1;i:46055;b:1;i:46110;b:1;i:46073;b:1;i:46124;b:1;i:46132;b:1;i:45983;b:1;i:46095;b:1;i:46136;b:1;i:45897;b:1;i:46162;b:1;i:46155;b:1;i:46161;b:1;i:46163;b:1;i:46092;b:1;i:46096;b:1;i:46145;b:1;i:46172;b:1;i:46151;b:1;i:46195;b:1;i:46202;b:1;i:46049;b:1;i:46050;b:1;i:46232;b:1;i:45945;b:1;i:46244;b:1;i:46234;b:1;i:46254;b:1;i:46230;b:1;i:46206;b:1;i:46174;b:1;i:46150;b:1;i:46268;b:1;i:46274;b:1;i:46271;b:1;i:46213;b:1;i:46253;b:1;i:46220;b:1;i:45674;b:1;i:45906;b:1;i:46282;b:1;i:46289;b:1;i:46286;b:1;i:46262;b:1;i:45949;b:1;i:45574;b:1;i:46112;b:1;i:46309;b:1;i:46034;b:1;i:46314;b:1;i:46318;b:1;i:46323;b:1;i:46018;b:1;i:46332;b:1;i:46333;b:1;i:46331;b:1;i:46281;b:1;i:46030;b:1;i:46364;b:1;i:46255;b:1;i:46367;b:1;i:46362;b:1;i:46378;b:1;i:46380;b:1;i:46390;b:1;i:46321;b:1;i:46393;b:1;i:46397;b:1;i:46335;b:1;i:46405;b:1;i:46249;b:1;i:46375;b:1;i:46423;b:1;i:46422;b:1;i:46421;b:1;i:46430;b:1;i:46402;b:1;i:46448;b:1;i:46322;b:1;i:46453;b:1;i:46379;b:1;i:46458;b:1;i:46045;b:1;i:46438;b:1;i:46342;b:1;i:46472;b:1;i:46476;b:1;i:46153;b:1;i:46486;b:1;i:46487;b:1;i:46488;b:1;i:46200;b:1;i:46499;b:1;i:46284;b:1;i:46496;b:1;i:46507;b:1;i:46356;b:1;i:46512;b:1;i:46475;b:1;i:46450;b:1;i:46266;b:1;i:46520;b:1;i:46494;b:1;i:46539;b:1;i:46533;b:1;i:46538;b:1;i:46540;b:1;i:46551;b:1;i:46555;b:1;i:46552;b:1;i:46452;b:1;i:46560;b:1;i:46535;b:1;i:46184;b:1;i:46127;b:1;i:46574;b:1;i:46291;b:1;i:46504;b:1;i:46484;b:1;i:46604;b:1;i:46605;b:1;i:46616;b:1;i:46619;b:1;i:46623;b:1;i:46624;b:1;i:46629;b:1;i:46628;b:1;i:46633;b:1;i:46635;b:1;i:46521;b:1;i:46652;b:1;i:46656;b:1;i:46657;b:1;i:46660;b:1;i:46658;b:1;i:46655;b:1;i:46639;b:1;i:46674;b:1;i:46676;b:1;i:46517;b:1;i:46626;b:1;i:46681;b:1;i:46561;b:1;i:46441;b:1;i:46695;b:1;i:46697;b:1;i:46631;b:1;i:46602;b:1;i:46528;b:1;i:46717;b:1;i:46723;b:1;i:46724;b:1;i:46736;b:1;i:46740;b:1;i:46734;b:1;i:46719;b:1;i:46241;b:1;i:46078;b:1;i:46750;b:1;i:46721;b:1;i:46641;b:1;i:46761;b:1;i:46764;b:1;i:46491;b:1;i:46778;b:1;i:46725;b:1;i:46727;b:1;i:46788;b:1;i:46706;b:1;i:46796;b:1;i:46801;b:1;i:46805;b:1;i:46806;b:1;i:46812;b:1;i:46804;b:1;i:46826;b:1;i:46689;b:1;i:46817;b:1;i:46832;b:1;i:46572;b:1;i:46830;b:1;i:46848;b:1;i:46851;b:1;i:46648;b:1;i:46861;b:1;i:46860;b:1;i:46857;b:1;i:46875;b:1;i:46876;b:1;i:46597;b:1;i:46838;b:1;i:46892;b:1;i:46888;b:1;i:46803;b:1;i:46885;b:1;i:46896;b:1;i:46897;b:1;i:46898;b:1;i:46904;b:1;i:46910;b:1;i:46922;b:1;i:46873;b:1;i:46929;b:1;i:46579;b:1;i:46593;b:1;i:46772;b:1;i:46931;b:1;i:46771;b:1;i:46915;b:1;i:46865;b:1;i:46959;b:1;i:46798;b:1;i:46917;b:1;i:46969;b:1;i:46968;b:1;i:46760;b:1;i:46418;b:1;i:44077;i:1;i:17560;i:0;i:46808;b:1;i:19375;i:1;i:17564;i:1;i:46886;b:1;i:46853;b:1;i:47001;b:1;i:47000;b:1;i:47005;b:1;i:47008;b:1;i:47004;b:1;i:47015;b:1;i:47016;b:1;i:47019;b:1;i:46998;b:1;i:46862;b:1;i:47028;b:1;i:46976;b:1;i:46912;b:1;i:47036;b:1;i:47038;b:1;i:46903;b:1;i:47039;b:1;i:47040;b:1;i:46794;b:1;i:47060;b:1;i:47011;b:1;i:46951;b:1;i:47045;b:1;i:47067;b:1;i:47076;b:1;i:46921;b:1;i:46615;b:1;i:47081;b:1;i:47104;b:1;i:47043;b:1;i:47002;b:1;i:46809;b:1;i:47125;b:1;i:47129;b:1;i:46957;b:1;i:47126;b:1;i:47080;b:1;i:47140;b:1;i:47141;b:1;i:47142;b:1;i:47130;b:1;i:47079;b:1;i:47037;b:1;i:46807;b:1;i:47143;b:1;i:47155;b:1;i:47159;b:1;i:47111;b:1;i:47164;b:1;i:47022;b:1;i:47108;b:1;i:47168;b:1;i:47175;b:1;i:47177;b:1;i:47106;b:1;i:47178;b:1;i:47179;b:1;i:47194;b:1;i:47192;b:1;i:47204;b:1;i:47216;b:1;i:47212;b:1;i:47214;b:1;i:47147;b:1;i:47217;b:1;i:47184;b:1;i:47228;b:1;i:47123;b:1;i:47197;b:1;i:47235;b:1;i:47176;b:1;i:47250;b:1;i:47260;b:1;i:47213;b:1;i:47146;b:1;i:47285;b:1;i:47284;b:1;i:47160;b:1;i:47172;b:1;i:47293;b:1;i:47294;b:1;i:47296;b:1;i:47299;b:1;i:47298;b:1;i:47300;b:1;i:47266;b:1;i:47261;b:1;i:47311;b:1;i:47313;b:1;i:47263;b:1;i:47323;b:1;i:47321;b:1;i:47223;b:1;i:47329;b:1;i:47334;b:1;i:47246;b:1;i:47338;b:1;i:47351;b:1;i:47356;b:1;i:47262;b:1;i:46996;b:1;i:47167;b:1;i:47374;b:1;i:47376;b:1;i:47391;b:1;i:47396;b:1;i:47371;b:1;i:47398;b:1;i:47310;b:1;i:47406;b:1;i:47189;b:1;i:47407;b:1;i:47412;b:1;i:47415;b:1;i:47416;b:1;i:47433;b:1;i:47434;b:1;i:47438;b:1;i:46943;b:1;i:47286;b:1;i:47445;b:1;i:47342;b:1;i:47449;b:1;i:47440;b:1;i:47446;b:1;i:47452;b:1;i:47276;b:1;i:47461;b:1;i:47460;b:1;i:47424;b:1;i:47471;b:1;i:47165;b:1;i:47479;b:1;i:47481;b:1;i:47480;b:1;i:46696;b:1;i:46625;b:1;i:47487;b:1;i:47490;b:1;i:47495;b:1;i:47496;b:1;i:47505;b:1;i:47506;b:1;i:47508;b:1;i:47510;b:1;i:47513;b:1;i:47291;b:1;i:47523;b:1;i:47501;b:1;i:47500;b:1;i:47448;b:1;i:47534;b:1;i:47543;b:1;i:47545;b:1;i:47522;b:1;i:47535;b:1;i:47553;b:1;i:47425;b:1;i:47561;b:1;i:47320;b:1;i:47567;b:1;i:47393;b:1;i:47573;b:1;i:47242;b:1;i:47572;b:1;i:47578;b:1;i:47585;b:1;i:47586;b:1;i:47560;b:1;i:47595;b:1;i:47593;b:1;i:47599;b:1;i:47527;b:1;i:47319;b:1;i:47602;b:1;i:47456;b:1;i:47436;b:1;i:47625;b:1;i:47630;b:1;i:47591;b:1;i:47648;b:1;i:47649;b:1;i:46713;b:1;i:47308;b:1;i:47352;b:1;i:47550;b:1;i:47350;b:1;i:47683;b:1;i:47691;b:1;i:47692;b:1;i:47469;b:1;i:47693;b:1;i:47710;b:1;i:47721;b:1;i:47731;b:1;i:47740;b:1;i:47739;b:1;i:47157;b:1;i:47758;b:1;i:47613;b:1;i:47563;b:1;i:47763;b:1;i:47772;b:1;i:47774;b:1;i:47778;b:1;i:47701;b:1;i:47791;b:1;i:47746;b:1;i:47682;b:1;i:47383;b:1;i:47802;b:1;i:47808;b:1;i:47819;b:1;i:47622;b:1;i:47830;b:1;i:47832;b:1;i:47834;b:1;i:47785;b:1;i:47845;b:1;i:47848;b:1;i:47831;b:1;i:47757;b:1;i:47715;b:1;i:47869;b:1;i:47780;b:1;i:47877;b:1;i:47875;b:1;i:47883;b:1;i:47896;i:1;i:47903;b:1;i:47906;b:1;i:47723;b:1;i:47866;b:1;i:47923;b:1;i:47925;b:1;i:47817;b:1;i:47937;b:1;i:47938;b:1;i:47936;b:1;i:47674;b:1;i:47947;b:1;i:47916;b:1;i:47959;i:0;i:47961;b:1;i:47839;b:1;i:47968;b:1;i:47876;b:1;i:47859;b:1;i:47811;b:1;i:47978;b:1;i:47962;b:1;i:47941;b:1;i:47984;b:1;i:47979;b:1;i:47993;b:1;i:47994;b:1;i:48004;b:1;i:47991;b:1;i:48018;b:1;i:47825;b:1;i:47844;b:1;i:48000;b:1;i:47977;b:1;i:47907;b:1;i:48040;b:1;i:48045;b:1;i:47868;b:1;i:48020;b:1;i:48053;b:1;i:48069;b:1;i:47849;b:1;i:47931;b:1;i:48043;b:1;i:48081;i:1;i:48089;b:1;i:48094;b:1;i:48093;b:1;i:48101;b:1;i:48098;b:1;i:47930;b:1;i:48107;b:1;i:48110;b:1;i:48032;b:1;i:48073;b:1;i:48137;b:1;i:48148;b:1;i:48138;b:1;i:48168;b:1;i:48125;b:1;i:48028;b:1;i:48175;b:1;i:48114;b:1;i:48182;b:1;i:48192;b:1;i:48172;b:1;i:48196;b:1;i:48177;b:1;i:48140;b:1;i:48131;b:1;i:48210;b:1;i:48190;b:1;i:48212;b:1;i:48208;b:1;i:48206;b:1;i:48213;b:1;i:48189;b:1;i:48217;b:1;i:48010;b:1;i:48236;b:1;i:47976;b:1;i:48237;b:1;i:48225;b:1;i:48263;i:1;i:45462;i:1;i:45463;i:1;i:48264;b:1;i:48227;b:1;i:48160;b:1;i:48166;b:1;i:48278;b:1;i:48281;b:1;i:48280;b:1;i:48283;b:1;i:48284;b:1;i:48294;b:1;i:48279;b:1;i:48297;b:1;i:48273;b:1;i:48169;b:1;i:48308;b:1;i:48293;b:1;i:47965;b:1;i:48327;b:1;i:48332;b:1;i:48328;b:1;i:48304;b:1;i:48348;b:1;i:48235;b:1;i:48115;b:1;i:48072;b:1;i:48317;b:1;i:48127;b:1;i:48335;b:1;i:48367;b:1;i:48218;b:1;i:47999;b:1;i:48215;b:1;i:48134;b:1;i:48385;b:1;i:48386;b:1;i:48296;b:1;i:48265;b:1;i:48146;b:1;i:48409;b:1;i:48411;b:1;i:48285;b:1;i:48047;b:1;i:48434;b:1;i:48388;b:1;i:47986;b:1;i:48391;b:1;i:48443;b:1;i:48480;b:1;i:48479;b:1;i:48221;b:1;i:48489;b:1;i:48445;b:1;i:48495;b:1;i:48494;b:1;i:48499;b:1;i:48230;b:1;i:48191;b:1;i:48511;b:1;i:48512;b:1;i:48534;b:1;i:48535;b:1;i:48536;b:1;i:48540;b:1;i:46877;i:1;i:48533;b:1;i:22317;b:1;i:16539;b:0;i:48561;b:1;i:15273;b:0;i:14084;b:0;i:48562;b:1;i:48397;b:1;i:48530;b:1;i:48568;b:1;i:48580;b:1;i:48582;b:1;i:48590;b:1;i:48543;b:1;i:48624;b:1;i:48625;b:1;i:48630;b:1;i:48484;b:1;i:48602;b:1;i:48623;b:1;i:48637;b:1;i:48633;b:1;i:48531;b:1;i:48651;b:1;i:48471;b:1;i:48525;b:1;i:48233;b:1;i:48313;b:1;i:48157;b:1;i:48642;b:1;i:48681;b:1;i:48648;b:1;i:48697;b:1;i:48527;b:1;i:48583;b:1;i:48539;b:1;i:48664;b:1;i:48719;b:1;i:48723;b:1;i:48729;b:1;i:48731;b:1;i:48702;b:1;i:48748;b:1;i:48751;b:1;i:48646;b:1;i:48692;b:1;i:48759;b:1;i:48725;b:1;i:48761;b:1;i:48753;b:1;i:48763;b:1;i:48705;b:1;i:48749;b:1;i:48326;b:1;i:48775;b:1;i:48778;b:1;i:48779;b:1;i:48770;b:1;i:48785;i:0;i:48790;b:1;i:48797;b:1;i:48706;b:1;i:48570;b:1;i:48815;b:1;i:48671;b:1;i:48732;b:1;i:48669;b:1;i:48333;b:1;i:48743;b:1;i:48814;b:1;i:48855;b:1;i:48836;b:1;i:48889;b:1;i:48895;b:1;i:48890;b:1;i:48904;b:1;i:48910;b:1;i:48747;b:1;i:48918;b:1;i:48925;b:1;i:48935;b:1;i:48700;b:1;i:48818;b:1;i:48947;b:1;i:48950;b:1;i:48941;b:1;i:48866;b:1;i:48903;b:1;i:48984;b:1;i:48990;b:1;i:48982;b:1;i:48972;b:1;i:49012;b:1;i:49018;b:1;i:48911;b:1;i:48983;b:1;i:48834;b:1;i:49050;b:1;i:48993;b:1;i:48820;b:1;i:48946;b:1;i:48958;b:1;i:48960;b:1;i:40301;i:1;i:49074;b:1;i:48995;b:1;i:49060;b:1;i:48970;b:1;i:23965;b:1;i:49102;b:1;i:49056;b:1;i:49053;b:1;i:49083;b:1;i:49085;b:1;i:49029;b:1;i:45828;i:1;i:47714;i:1;i:49046;b:1;i:49106;b:1;i:49134;b:1;i:49132;b:1;i:49131;b:1;i:49140;b:1;i:49143;b:1;i:49104;b:1;i:48802;b:1;i:49162;b:1;i:49163;b:1;i:49166;b:1;i:49087;b:1;i:49169;b:1;i:49159;b:1;i:49172;b:1;i:49081;b:1;i:49197;b:1;i:49198;b:1;i:49139;b:1;i:49194;b:1;i:49182;b:1;i:49204;b:1;i:49206;b:1;i:49209;b:1;i:49220;b:1;i:49216;b:1;i:49211;b:1;i:49226;b:1;i:49238;b:1;i:49248;b:1;i:49249;b:1;i:49170;b:1;i:49219;b:1;i:49224;b:1;i:49246;b:1;i:49250;b:1;i:49147;b:1;i:49260;b:1;i:49125;b:1;i:49277;b:1;i:49122;b:1;i:49294;b:1;i:48721;b:1;i:49191;b:1;i:49301;b:1;i:49168;b:1;i:49281;b:1;i:49315;b:1;i:49161;b:1;i:49317;b:1;i:49330;b:1;i:49327;b:1;i:49344;b:1;i:49265;b:1;i:49348;b:1;i:49349;b:1;i:49310;b:1;i:49359;b:1;i:49270;b:1;i:49269;b:1;i:49341;b:1;i:49337;b:1;i:49376;b:1;i:49183;b:1;i:49381;b:1;i:49384;b:1;i:49368;b:1;i:49253;b:1;i:49361;b:1;i:49404;b:1;i:49392;b:1;i:49396;b:1;i:49407;b:1;i:49370;b:1;i:49447;b:1;i:49450;b:1;i:49429;b:1;i:49379;b:1;i:49117;b:1;i:49366;b:1;i:49481;b:1;i:49154;b:1;i:49492;b:1;i:49506;b:1;i:49354;b:1;i:49207;b:1;i:49520;b:1;i:49522;b:1;i:49524;b:1;i:49511;b:1;i:49454;b:1;i:49483;b:1;i:49514;b:1;i:49515;b:1;i:49488;b:1;i:49540;b:1;i:49440;b:1;i:49546;i:0;i:49547;i:0;i:49548;b:1;i:49434;b:1;i:49553;b:1;i:49230;b:1;i:49123;b:1;i:49555;b:1;i:49565;b:1;i:49568;b:1;i:49552;b:1;i:49456;b:1;i:49588;b:1;i:49572;b:1;i:49597;b:1;i:49578;b:1;i:49517;b:1;i:49603;b:1;i:49537;b:1;i:49611;b:1;i:49437;b:1;i:49621;b:1;i:49630;b:1;i:49634;i:0;i:49633;i:0;i:49635;i:0;i:49638;b:1;i:49639;b:1;i:49647;b:1;i:49605;b:1;i:49607;b:1;i:49609;b:1;i:49636;b:1;i:49661;b:1;i:49664;b:1;i:49666;b:1;i:49668;b:1;i:49663;b:1;i:49463;b:1;i:49691;b:1;i:49684;b:1;i:49711;b:1;i:49707;b:1;i:49646;b:1;i:49686;b:1;i:49712;b:1;i:49718;i:0;i:49721;b:1;i:49741;b:1;i:49745;b:1;i:49616;b:1;i:49752;b:1;i:49756;b:1;i:49762;b:1;i:49767;b:1;i:49774;b:1;i:49764;b:1;i:49765;b:1;i:49792;b:1;i:49743;b:1;i:49771;b:1;i:49797;b:1;i:49682;b:1;i:49679;b:1;i:49115;b:1;i:49726;b:1;i:49769;b:1;i:49809;b:1;i:49813;b:1;i:49812;b:1;i:49810;b:1;i:49814;b:1;i:49785;b:1;i:49835;b:1;i:49847;b:1;i:49719;b:1;i:49701;b:1;i:49589;b:1;i:49748;b:1;i:49791;b:1;i:49799;b:1;i:49876;b:1;i:49831;b:1;i:49894;b:1;i:49899;b:1;i:49902;b:1;i:49868;b:1;i:49884;b:1;i:49787;b:1;i:49855;b:1;i:49913;b:1;i:49898;b:1;i:49919;b:1;i:49921;b:1;i:49922;b:1;i:49929;b:1;i:49912;b:1;i:49942;b:1;i:49947;b:1;i:49948;b:1;i:49889;b:1;i:49723;b:1;i:49863;b:1;i:49740;b:1;i:49914;b:1;i:49975;i:0;i:49977;b:1;i:49981;i:1;i:49958;b:1;i:49983;b:1;i:49970;b:1;i:49893;b:1;i:49964;b:1;i:49963;b:1;i:49976;i:0;i:50017;b:1;i:49932;b:1;i:50009;b:1;i:50007;b:1;i:49965;b:1;i:50027;b:1;i:49992;b:1;i:49972;b:1;i:50029;b:1;i:50042;b:1;i:50019;b:1;i:50077;b:1;i:50078;b:1;i:50079;b:1;i:50053;b:1;i:50106;b:1;i:49660;b:1;i:49662;b:1;i:49667;b:1;i:49669;b:1;i:49671;b:1;i:49673;b:1;i:49675;b:1;i:49690;b:1;i:49793;b:1;i:49816;b:1;i:50048;b:1;i:50123;b:1;i:50125;b:1;i:50059;b:1;i:50142;b:1;i:50156;b:1;i:50168;b:1;i:50169;b:1;i:50190;b:1;i:49985;b:1;i:50022;b:1;i:50200;b:1;i:50138;b:1;i:50202;b:1;i:50216;b:1;i:50167;b:1;i:50247;b:1;i:50254;b:1;i:50258;b:1;i:50157;b:1;i:50256;b:1;i:50175;b:1;i:50045;b:1;i:50084;b:1;i:50218;b:1;i:50231;b:1;i:50204;b:1;i:50181;b:1;i:50182;b:1;i:50243;b:1;i:50096;b:1;i:50298;b:1;i:50064;b:1;i:50344;b:1;i:50055;b:1;i:50364;b:1;i:50387;b:1;i:50392;b:1;i:50397;b:1;i:50404;b:1;i:50325;b:1;i:50409;b:1;i:50356;b:1;i:50279;b:1;i:50426;b:1;i:50390;b:1;i:50406;b:1;i:50372;b:1;i:50427;b:1;i:50016;b:1;i:50332;b:1;i:50147;b:1;i:50417;b:1;i:50436;b:1;i:50438;b:1;i:38040;i:1;i:50402;b:1;i:50459;b:1;i:50456;b:1;i:50240;b:1;i:50435;b:1;i:50088;b:1;i:50375;b:1;i:50354;b:1;i:50312;b:1;i:50492;b:1;i:50519;b:1;i:50526;b:1;i:50379;b:1;i:50533;b:1;i:50537;b:1;i:50026;b:1;i:50551;b:1;i:50549;b:1;i:50295;b:1;i:50363;b:1;i:50570;b:1;i:50542;b:1;i:50049;b:1;i:50564;b:1;i:50593;b:1;i:50431;b:1;i:49811;b:1;i:50489;b:1;i:50555;b:1;i:50597;b:1;i:50623;b:1;i:50560;b:1;i:50635;b:1;i:50641;b:1;i:50653;b:1;i:50663;b:1;i:50637;b:1;i:50678;b:1;i:50677;b:1;i:50693;b:1;i:50367;b:1;i:50627;b:1;i:50619;b:1;i:50658;b:1;i:50622;b:1;i:50723;b:1;i:50736;b:1;i:50733;b:1;i:50750;b:1;i:50683;b:1;i:50581;i:1;i:50777;b:1;i:50701;b:1;i:50691;b:1;i:50785;b:1;i:50787;b:1;i:50789;b:1;i:50794;b:1;i:50793;b:1;i:50790;b:1;i:50802;b:1;i:50778;b:1;i:50807;b:1;i:50782;b:1;i:50828;b:1;i:50774;b:1;i:50845;b:1;i:50521;b:1;i:50642;b:1;i:50652;b:1;i:50507;b:1;i:50798;b:1;i:50810;b:1;i:50871;b:1;i:50886;b:1;i:50887;b:1;i:50888;b:1;i:50893;b:1;i:50656;b:1;i:50904;b:1;i:50849;b:1;i:50840;b:1;i:50935;b:1;i:50941;b:1;i:50901;b:1;i:50873;b:1;i:50881;b:1;i:50800;b:1;i:50738;b:1;i:50946;b:1;i:50804;b:1;i:50930;b:1;i:50928;b:1;i:50958;b:1;i:50976;b:1;i:50972;b:1;i:50879;b:1;i:50591;b:1;i:50648;b:1;i:50989;b:1;i:50898;b:1;i:50933;b:1;i:50876;b:1;i:50982;b:1;i:51006;b:1;i:51017;b:1;i:51023;b:1;i:51024;b:1;i:51000;b:1;i:50949;b:1;i:51029;b:1;i:51031;b:1;i:51027;b:1;i:51052;b:1;i:51002;b:1;i:51054;b:1;i:51022;b:1;i:51073;b:1;i:51042;b:1;i:51047;b:1;i:51063;b:1;i:51064;b:1;i:51082;b:1;i:51104;b:1;i:50978;b:1;i:51065;b:1;i:51128;b:1;i:51148;b:1;i:51132;b:1;i:51143;b:1;i:51135;b:1;i:51138;b:1;i:51163;b:1;i:51095;b:1;i:51191;b:1;i:51173;b:1;i:51195;b:1;i:51184;b:1;i:51162;b:1;i:51182;b:1;i:51150;b:1;i:51089;b:1;i:51120;b:1;i:51217;b:1;i:51225;b:1;i:51211;b:1;i:51224;b:1;i:51014;b:1;i:51237;b:1;i:51249;b:1;i:51246;i:1;i:45513;i:1;i:51239;b:1;i:51250;b:1;i:51251;b:1;i:51261;b:1;i:51258;b:1;i:51283;b:1;i:51285;b:1;i:51284;b:1;i:51292;b:1;i:51248;b:1;i:51297;b:1;i:51302;b:1;i:51299;b:1;i:51300;b:1;i:51304;b:1;i:51311;b:1;i:51312;b:1;i:51266;b:1;i:51324;b:1;i:51276;b:1;i:51279;b:1;i:51338;b:1;i:51350;b:1;i:51318;b:1;i:51355;b:1;i:51321;b:1;i:51260;b:1;i:51327;b:1;i:51363;b:1;i:51371;b:1;i:51373;b:1;i:51388;b:1;i:51389;b:1;i:51390;b:1;i:51395;b:1;i:51394;b:1;i:51370;b:1;i:51403;b:1;i:51402;b:1;i:51349;b:1;i:51289;b:1;i:51421;b:1;i:51310;b:1;i:51434;b:1;i:51420;b:1;i:51435;b:1;i:51440;b:1;i:51442;b:1;i:51447;b:1;i:51449;b:1;i:51454;b:1;i:51456;b:1;i:51461;b:1;i:51471;b:1;i:51452;b:1;i:51341;b:1;i:51462;b:1;i:51315;b:1;i:51487;b:1;i:51498;b:1;i:51505;b:1;i:51520;b:1;i:51526;b:1;i:51530;b:1;i:51531;b:1;i:51532;b:1;i:51497;b:1;i:51439;b:1;i:51436;b:1;i:51528;b:1;i:51537;b:1;i:51548;b:1;i:51570;b:1;i:51486;b:1;i:51585;b:1;i:51432;b:1;i:51573;b:1;i:51595;b:1;i:51481;b:1;i:51534;b:1;i:51502;b:1;i:51495;b:1;i:51480;b:1;i:51382;b:1;i:51576;b:1;i:51594;b:1;i:51609;b:1;i:51612;b:1;i:51599;b:1;i:51545;b:1;i:51607;b:1;i:51637;b:1;i:51638;b:1;i:51640;b:1;i:51641;b:1;i:51631;b:1;i:51562;b:1;i:51720;b:1;i:51724;b:1;i:51636;b:1;i:51729;b:1;i:51579;b:1;i:51670;b:1;i:51733;b:1;i:51764;b:1;i:51716;b:1;i:51767;b:1;i:51775;b:1;i:51776;b:1;i:51797;b:1;i:51794;b:1;i:51718;b:1;i:51813;b:1;i:51697;b:1;i:51698;b:1;i:51707;b:1;i:51706;b:1;i:51841;b:1;i:51851;b:1;i:51818;b:1;i:51862;b:1;i:51866;b:1;i:51774;b:1;i:51778;b:1;i:51888;b:1;i:51611;b:1;i:28701;i:0;i:51898;b:1;i:51785;b:1;i:51909;b:1;i:51917;b:1;i:51871;b:1;i:51874;b:1;i:51873;b:1;i:51879;b:1;i:51897;b:1;i:51936;b:1;i:51907;b:1;i:51939;b:1;i:51942;b:1;i:51937;b:1;i:51930;b:1;i:51722;b:1;i:51952;b:1;i:51959;b:1;i:51944;b:1;i:51927;b:1;i:51926;b:1;i:51961;b:1;i:51962;b:1;i:51914;b:1;i:51954;b:1;i:51951;b:1;i:51988;b:1;i:51949;b:1;i:51996;b:1;i:51881;b:1;i:52013;b:1;i:52015;b:1;i:52017;b:1;i:52020;b:1;i:52025;b:1;i:52042;b:1;i:52043;b:1;i:52018;b:1;i:52057;b:1;i:52058;b:1;i:52065;b:1;i:52022;b:1;i:52071;b:1;i:52085;b:1;i:52095;b:1;i:52092;b:1;i:52097;b:1;i:52123;b:1;i:52124;b:1;i:52116;b:1;i:52136;b:1;i:52140;b:1;i:52063;b:1;i:51529;b:1;i:51527;b:1;i:52120;b:1;i:52122;b:1;i:52155;b:1;i:52150;b:1;i:52162;b:1;i:52157;b:1;i:52106;b:1;i:52115;b:1;i:52167;b:1;i:52113;b:1;i:51981;b:1;i:52220;b:1;i:52070;b:1;i:51807;b:1;i:51935;b:1;i:52228;b:1;i:52237;b:1;i:52235;b:1;i:52266;b:1;i:52161;b:1;i:52214;b:1;i:52219;b:1;i:52285;b:1;i:52111;b:1;i:52306;b:1;i:52276;b:1;i:52148;b:1;i:52328;b:1;i:52329;b:1;i:52330;b:1;i:52294;b:1;i:52358;b:1;i:52369;b:1;i:52154;b:1;i:52372;b:1;i:52180;b:1;i:52393;b:1;i:52392;b:1;i:52396;b:1;i:52298;b:1;i:52419;b:1;i:52386;b:1;i:52424;b:1;i:52425;b:1;i:52437;b:1;i:52448;b:1;i:52449;b:1;i:52394;b:1;i:52455;b:1;i:52474;b:1;i:52451;b:1;i:52543;b:1;i:52556;b:1;i:52560;b:1;i:52375;b:1;i:52561;b:1;i:52569;b:1;i:52573;b:1;i:52540;b:1;i:52599;b:1;i:52606;b:1;i:52596;b:1;i:52559;b:1;i:52558;b:1;i:52334;b:1;i:52602;b:1;i:52624;b:1;i:52315;b:1;i:52537;b:1;i:52623;b:1;i:52645;b:1;i:52535;b:1;i:52661;b:1;i:52664;b:1;i:52666;b:1;i:52670;b:1;i:52672;b:1;i:52675;b:1;i:52684;b:1;i:52662;b:1;i:52660;b:1;i:52696;b:1;i:52325;b:1;i:52702;b:1;i:52709;b:1;i:52705;b:1;i:52710;b:1;i:52711;b:1;i:52719;b:1;i:52721;b:1;i:51963;i:1;i:52724;i:0;i:52674;b:1;i:52734;b:1;i:52732;b:1;i:52738;b:1;i:52765;b:1;i:52773;b:1;i:52775;b:1;i:52747;b:1;i:52743;b:1;i:52777;b:1;i:52763;b:1;i:52797;b:1;i:52800;b:1;i:52801;b:1;i:52802;b:1;i:52740;b:1;i:52811;b:1;i:52779;b:1;i:52799;b:1;i:52821;b:1;i:52826;b:1;i:52768;b:1;i:52845;b:1;i:52847;b:1;i:52848;b:1;i:52855;b:1;i:52687;b:1;i:52604;b:1;i:52752;b:1;i:52806;b:1;i:52669;b:1;i:52881;b:1;i:52735;b:1;i:52782;b:1;i:52892;b:1;i:52895;b:1;i:52897;b:1;i:52910;b:1;i:52909;b:1;i:52890;b:1;i:52769;b:1;i:52917;b:1;i:52825;b:1;i:52944;b:1;i:52945;b:1;i:52946;b:1;i:52937;b:1;i:52914;b:1;i:52966;b:1;i:52962;b:1;i:52919;b:1;i:52790;b:1;i:52961;b:1;i:19071;i:1;i:52980;i:0;i:52838;b:1;i:52967;b:1;i:52984;b:1;i:52860;b:1;i:52947;b:1;i:52952;b:1;i:52939;b:1;i:53019;b:1;i:53031;b:1;i:53036;b:1;i:53040;b:1;i:53042;b:1;i:53048;b:1;i:53049;b:1;i:53051;b:1;i:53050;b:1;i:52988;b:1;i:52791;b:1;i:53058;b:1;i:52976;b:1;i:53027;b:1;i:53070;b:1;i:53087;b:1;i:53083;b:1;i:53096;b:1;i:53053;b:1;i:53094;b:1;i:53111;b:1;i:53110;b:1;i:53120;b:1;i:53137;b:1;i:53118;b:1;i:53082;b:1;i:53148;b:1;i:53150;b:1;i:53147;b:1;i:53158;b:1;i:53162;b:1;i:53163;b:1;i:53164;b:1;i:53177;b:1;i:53127;b:1;i:53183;b:1;i:53185;b:1;i:53187;b:1;i:53102;b:1;i:53022;b:1;i:53216;b:1;i:53208;b:1;i:53075;b:1;i:53234;b:1;i:53228;b:1;i:53237;b:1;i:53202;b:1;i:53192;b:1;i:53248;b:1;i:53251;b:1;i:53253;b:1;i:53269;b:1;i:53275;b:1;i:53273;b:1;i:53278;b:1;i:53280;b:1;i:53287;b:1;i:53288;b:1;i:53200;b:1;i:53302;b:1;i:53264;b:1;i:53313;b:1;i:53317;b:1;i:53303;b:1;i:53311;b:1;i:53333;b:1;i:53338;b:1;i:53329;b:1;i:53354;b:1;i:53361;b:1;i:53366;b:1;i:53368;b:1;i:53367;b:1;i:53339;b:1;i:53353;b:1;i:53352;b:1;i:53401;b:1;i:53383;b:1;i:53400;b:1;i:53435;b:1;i:53437;b:1;i:53444;b:1;i:53439;b:1;i:53455;b:1;i:53459;b:1;i:53324;b:1;i:53460;b:1;i:53465;b:1;i:53438;b:1;i:53456;b:1;i:53402;b:1;i:53497;b:1;i:53480;b:1;i:53494;b:1;i:53505;b:1;i:53510;b:1;i:53515;b:1;i:53509;b:1;i:53537;b:1;i:53536;b:1;i:53542;b:1;i:53543;b:1;i:53549;b:1;i:53556;b:1;i:52400;b:1;i:53559;b:1;i:53574;b:1;i:53582;b:1;i:53579;b:1;i:53551;b:1;i:53562;b:1;i:53583;b:1;i:53557;b:1;i:53565;b:1;i:53577;b:1;i:53591;b:1;i:53590;b:1;i:53593;b:1;i:53596;b:1;i:53594;b:1;i:53613;b:1;i:53575;b:1;i:53617;b:1;i:53586;b:1;i:53639;b:1;i:53544;b:1;i:53636;i:0;i:53629;b:1;i:53605;b:1;i:53621;b:1;i:53659;b:1;i:53660;b:1;i:53662;b:1;i:53661;b:1;i:53643;b:1;i:53657;b:1;i:53677;b:1;i:52907;i:1;i:53689;b:1;i:53697;b:1;i:53701;b:1;i:53560;b:1;i:53700;b:1;i:53702;b:1;i:53705;b:1;i:53704;b:1;i:53696;b:1;i:53709;b:1;i:53727;b:1;i:53731;b:1;i:53734;b:1;i:53669;b:1;i:53675;b:1;i:53653;b:1;i:53645;b:1;i:53644;b:1;i:53746;b:1;i:53753;b:1;i:53752;b:1;i:53755;b:1;i:53754;b:1;i:53761;b:1;i:53769;b:1;i:53768;b:1;i:53772;b:1;i:53773;b:1;i:53775;b:1;i:53778;b:1;i:53776;b:1;i:53781;b:1;i:53779;b:1;i:53786;i:0;i:53787;i:0;i:53788;i:0;i:53790;b:1;i:53735;b:1;i:53691;b:1;i:53673;b:1;i:53668;b:1;i:53647;b:1;i:53791;b:1;i:53794;b:1;i:53805;i:0;i:53806;i:0;i:53808;i:0;i:53809;i:0;i:53810;i:0;i:53667;b:1;i:53722;b:1;i:53682;b:1;i:53839;b:1;i:53855;b:1;i:53838;b:1;i:53831;b:1;i:53871;b:1;i:53870;b:1;i:53833;b:1;i:53867;b:1;i:53860;b:1;i:53840;b:1;i:53835;b:1;i:53862;b:1;i:53873;b:1;i:53834;b:1;i:53882;b:1;i:53893;b:1;i:53894;b:1;i:53892;b:1;i:53878;b:1;i:53209;b:1;i:53863;b:1;i:53903;b:1;i:53905;b:1;i:53916;b:1;i:53606;b:1;i:53607;b:1;i:53642;b:1;i:53930;b:1;i:53932;b:1;i:53928;b:1;i:53911;b:1;i:53910;b:1;i:53906;b:1;i:53884;b:1;i:53943;b:1;i:53944;b:1;i:53914;b:1;i:53872;b:1;i:53960;b:1;i:53958;b:1;i:53962;b:1;i:53976;b:1;i:53964;b:1;i:53890;b:1;i:53981;b:1;i:53995;b:1;i:53987;b:1;i:54006;b:1;i:54002;b:1;i:54009;b:1;i:54013;b:1;i:53997;b:1;i:54018;b:1;i:54019;b:1;i:54017;b:1;i:54025;b:1;i:54026;b:1;i:53933;b:1;i:54032;b:1;i:54004;b:1;i:53992;b:1;i:53991;b:1;i:54003;b:1;i:54035;b:1;i:54029;b:1;i:54012;b:1;i:54052;b:1;i:54054;b:1;i:54047;b:1;i:54055;b:1;i:54044;b:1;i:54058;b:1;i:54051;b:1;i:54063;b:1;i:54049;b:1;i:54068;b:1;i:54071;b:1;i:54060;b:1;i:54057;b:1;i:54053;b:1;i:54085;b:1;i:54090;b:1;i:54064;b:1;i:54097;b:1;i:54048;b:1;i:54103;b:1;i:54031;b:1;i:54078;b:1;i:54022;b:1;i:54129;b:1;i:54130;b:1;i:54131;b:1;i:54132;b:1;i:54139;b:1;i:54110;b:1;i:54138;b:1;i:54124;b:1;i:54143;b:1;i:54142;b:1;i:54154;b:1;i:54136;b:1;i:54179;b:1;i:54184;b:1;i:54192;b:1;i:54193;b:1;i:54188;b:1;i:54194;b:1;i:54168;b:1;i:54162;b:1;i:54133;b:1;i:54214;b:1;i:54195;b:1;i:54217;b:1;i:54218;b:1;i:54219;b:1;i:54223;b:1;i:54220;b:1;i:54234;b:1;i:54232;b:1;i:54231;b:1;i:54241;b:1;i:54242;b:1;i:54248;b:1;i:54250;b:1;i:54251;b:1;i:54252;b:1;i:54255;b:1;i:54254;b:1;i:54249;b:1;i:54257;b:1;i:54216;b:1;i:54153;b:1;i:54270;b:1;i:54258;b:1;i:54010;b:1;i:54279;b:1;i:54165;b:1;i:54287;b:1;i:54291;b:1;i:54180;b:1;i:54299;b:1;i:54288;b:1;i:54307;b:1;i:54308;b:1;i:54313;b:1;i:54303;b:1;i:54311;b:1;i:54323;b:1;i:54325;b:1;i:54333;b:1;i:54343;b:1;i:54345;b:1;i:54347;b:1;i:54348;b:1;i:54353;b:1;i:54328;b:1;i:54354;b:1;i:54355;b:1;i:54356;b:1;i:54362;b:1;i:54334;b:1;i:54337;b:1;i:53980;b:1;i:54363;b:1;i:54359;b:1;i:54371;b:1;i:54372;b:1;i:54336;b:1;i:54382;b:1;i:54368;b:1;i:54385;b:1;i:54388;b:1;i:54403;b:1;i:54389;b:1;i:54411;b:1;i:53792;b:1;i:53793;b:1;i:53798;b:1;i:53799;b:1;i:53827;b:1;i:54452;b:1;i:54407;b:1;i:54459;b:1;i:54413;b:1;i:54471;b:1;i:54475;b:1;i:54437;b:1;i:54478;b:1;i:54470;b:1;i:54472;b:1;i:54480;b:1;i:54466;b:1;i:54423;b:1;i:54490;b:1;i:54484;b:1;i:54454;b:1;i:54495;b:1;i:54494;b:1;i:54473;b:1;i:54503;b:1;i:54509;b:1;i:54513;b:1;i:54514;b:1;i:54448;b:1;i:54519;b:1;i:54520;b:1;i:54527;b:1;i:54542;b:1;i:54545;b:1;i:54546;b:1;i:54548;b:1;i:54506;b:1;i:54541;b:1;i:54552;b:1;i:54555;b:1;i:54554;b:1;i:54558;b:1;i:54568;b:1;i:54569;b:1;i:54567;b:1;i:54580;b:1;i:54586;b:1;i:54589;b:1;i:54605;b:1;i:54600;b:1;i:54585;b:1;i:54611;b:1;i:54610;b:1;i:54613;b:1;i:54630;b:1;i:54619;b:1;i:54590;b:1;i:54574;b:1;i:54632;b:1;i:54657;b:1;i:54658;i:0;i:54667;b:1;i:54666;b:1;i:54676;b:1;i:54674;b:1;i:54689;b:1;i:54694;b:1;i:54702;b:1;i:54709;b:1;i:54655;b:1;i:54711;b:1;i:54644;b:1;i:54726;b:1;i:54725;b:1;i:54665;b:1;i:54731;b:1;i:54739;b:1;i:54737;b:1;i:54721;b:1;i:54692;b:1;i:54662;b:1;i:54747;b:1;i:54748;b:1;i:54754;b:1;i:54758;b:1;i:54761;b:1;i:54762;b:1;i:54766;b:1;i:54769;b:1;i:54736;b:1;i:54774;b:1;i:54777;b:1;i:54782;b:1;i:54787;b:1;i:54778;b:1;i:54727;b:1;i:54797;b:1;i:54801;b:1;i:54808;b:1;i:54823;b:1;i:54826;b:1;i:54806;b:1;i:54804;b:1;i:54785;b:1;i:54776;b:1;i:54824;b:1;i:54819;b:1;i:54817;b:1;i:54814;b:1;i:54841;b:1;i:54845;b:1;i:54812;b:1;i:54850;b:1;i:54849;b:1;i:54847;b:1;i:54860;b:1;i:53684;i:1;i:54868;b:1;i:54873;b:1;i:54894;b:1;i:54911;b:1;i:54919;b:1;i:54922;b:1;i:54925;b:1;i:54859;b:1;i:54926;b:1;i:54931;b:1;i:54933;b:1;i:54944;b:1;i:54951;b:1;i:54972;b:1;i:54974;b:1;i:54983;b:1;i:54987;b:1;i:54991;b:1;i:54995;b:1;i:54989;b:1;i:54973;b:1;i:54997;b:1;i:55017;b:1;i:54928;b:1;i:55049;b:1;i:55050;b:1;i:55054;b:1;i:55058;b:1;i:55062;b:1;i:55069;b:1;i:55052;b:1;i:54369;b:1;i:55093;b:1;i:55044;b:1;i:55085;b:1;i:55098;b:1;i:55099;b:1;i:55101;b:1;i:55090;b:1;i:55106;b:1;i:55112;b:1;i:55006;b:1;i:55115;b:1;i:55104;b:1;i:55120;b:1;i:55132;b:1;i:55015;b:1;i:55105;b:1;i:55173;b:1;i:55155;b:1;i:55188;i:1;i:55083;b:1;i:55194;b:1;i:55197;b:1;i:55110;b:1;i:55187;b:1;i:55030;b:1;i:55169;b:1;i:55165;b:1;i:55222;b:1;i:55228;b:1;i:55220;b:1;i:55227;b:1;i:55224;b:1;i:55231;b:1;i:55237;b:1;i:55240;b:1;i:55235;b:1;i:55243;b:1;i:55246;b:1;i:55248;b:1;i:55236;b:1;i:55249;b:1;i:55254;b:1;i:55257;b:1;i:55230;b:1;i:55260;b:1;i:55262;b:1;i:55123;b:1;i:55266;b:1;i:55268;b:1;i:55259;b:1;i:55276;b:1;i:55261;b:1;i:55279;b:1;i:55281;i:1;i:54861;i:1;i:55275;b:1;i:55303;i:1;i:55331;b:1;i:55272;b:1;i:55287;b:1;i:55363;b:1;i:55368;b:1;i:55374;b:1;i:55375;b:1;i:55380;b:1;i:55390;b:1;i:55332;b:1;i:55393;b:1;i:55370;b:1;i:55397;b:1;i:55399;b:1;i:53655;b:1;i:55401;b:1;i:55403;b:1;i:55413;b:1;i:55415;b:1;i:55417;b:1;i:55420;b:1;i:55422;b:1;i:55424;b:1;i:55423;b:1;i:55421;b:1;i:55440;b:1;i:55326;b:1;i:55446;b:1;i:55445;b:1;i:55453;b:1;i:55432;b:1;i:55448;b:1;i:55458;b:1;i:55460;b:1;i:55464;b:1;i:55439;b:1;i:55461;b:1;i:55435;b:1;i:55477;b:1;i:55480;b:1;i:55491;b:1;i:55492;b:1;i:55511;b:1;i:55508;b:1;i:55521;b:1;i:55525;b:1;i:55541;b:1;i:55532;b:1;i:55551;b:1;i:55554;b:1;i:55561;b:1;i:54111;b:1;i:55565;b:1;i:55571;b:1;i:55574;b:1;i:55577;b:1;i:55576;b:1;i:55575;b:1;i:55582;b:1;i:55584;b:1;i:55588;b:1;i:55585;b:1;i:55540;b:1;i:55602;b:1;i:55604;b:1;i:55621;b:1;i:55617;b:1;i:55534;b:1;i:55653;b:1;i:55655;b:1;i:55654;b:1;i:55624;b:1;i:55649;b:1;i:55645;b:1;i:55667;b:1;i:55668;b:1;i:55644;b:1;i:55671;b:1;i:55683;b:1;i:55688;b:1;i:55596;b:1;i:55697;b:1;i:55699;b:1;i:55660;b:1;i:55720;b:1;i:55600;b:1;i:55728;b:1;i:55726;b:1;i:55731;b:1;i:55710;b:1;i:55718;b:1;i:55757;b:1;i:55759;b:1;i:55755;b:1;i:55750;b:1;i:55760;b:1;i:55754;b:1;i:55769;b:1;i:55721;b:1;i:55712;b:1;i:55765;b:1;i:55799;b:1;i:55801;b:1;i:55819;b:1;i:55795;b:1;i:55839;b:1;i:55844;b:1;i:55858;b:1;i:55874;b:1;i:55865;b:1;i:55892;b:1;i:55863;b:1;i:55920;b:1;i:55928;b:1;i:55929;b:1;i:55935;b:1;i:55942;b:1;i:55944;b:1;i:55952;b:1;i:55965;b:1;i:55927;b:1;i:55981;b:1;i:55982;b:1;i:55975;b:1;i:55943;b:1;i:55993;b:1;i:55997;b:1;i:55848;b:1;i:56007;b:1;i:56010;b:1;i:56011;b:1;i:56024;b:1;i:56027;b:1;i:56020;b:1;i:56031;b:1;i:56014;i:0;i:55716;i:1;i:55715;i:1;i:56042;b:1;i:56045;b:1;i:56043;b:1;i:56048;b:1;i:56028;b:1;i:56054;b:1;i:56064;b:1;i:56068;b:1;i:56066;b:1;i:56084;b:1;i:56088;b:1;i:56091;b:1;i:56098;b:1;i:56074;b:1;i:56099;b:1;i:56065;b:1;i:56108;b:1;i:56094;b:1;i:56120;b:1;i:56114;b:1;i:56053;b:1;i:56132;b:1;i:56131;b:1;i:45281;i:1;i:56136;b:1;i:56151;b:1;i:56152;b:1;i:56155;b:1;i:56154;b:1;i:56141;b:1;i:56156;b:1;i:56144;b:1;i:56150;b:1;i:56097;b:1;i:56148;b:1;i:56179;b:1;i:56186;b:1;i:56187;b:1;i:56185;b:1;i:56188;b:1;i:56129;b:1;i:56134;b:1;i:56209;b:1;i:56177;b:1;i:56228;b:1;i:56216;b:1;i:56237;b:1;i:56226;b:1;i:56240;b:1;i:56245;b:1;i:56175;b:1;i:56247;b:1;i:56264;b:1;i:56265;b:1;i:56268;b:1;i:56254;b:1;i:56255;b:1;i:56276;b:1;i:56275;b:1;i:56270;b:1;i:56293;b:1;i:56297;b:1;i:56299;i:1;i:56292;b:1;i:56304;b:1;i:56309;i:0;i:55717;i:1;i:56315;i:1;i:56320;b:1;i:56319;b:1;i:56322;b:1;i:56326;b:1;i:56325;b:1;i:56327;b:1;i:56310;b:1;i:56337;b:1;i:56339;b:1;i:56350;b:1;i:56360;b:1;i:56362;b:1;i:56363;b:1;i:56356;b:1;i:56357;b:1;i:56366;i:1;i:56354;i:1;i:56333;i:1;i:56318;i:1;i:56312;i:1;i:56305;i:1;i:56285;i:1;i:56284;i:1;i:56282;i:1;i:56369;b:1;i:56273;i:1;i:56272;i:1;i:56182;i:1;i:56205;i:0;i:56168;i:1;i:56370;b:1;i:56110;i:1;i:56367;b:1;i:56385;b:1;i:56386;b:1;i:56387;i:1;i:56388;i:1;i:56391;b:1;i:56390;b:1;i:56395;b:1;i:56396;b:1;i:56405;b:1;i:56406;b:1;i:56436;b:1;i:56443;b:1;i:56444;b:1;i:56445;b:1;i:51378;b:1;i:56460;i:1;i:56435;i:1;i:56462;b:1;i:56469;b:1;i:56452;b:1;i:56477;b:1;i:56472;b:1;i:56487;b:1;i:56488;b:1;i:56491;b:1;i:56478;b:1;i:56461;b:1;i:56494;b:1;i:56480;b:1;i:56500;b:1;i:56504;b:1;i:56471;i:1;i:56499;i:1;i:56493;i:0;i:56450;i:0;i:56438;i:0;i:56409;i:1;i:56408;i:1;i:56510;b:1;i:56511;b:1;i:56515;b:1;i:56517;b:1;i:56522;b:1;i:56523;b:1;i:56525;b:1;i:56524;b:1;i:56527;b:1;i:56505;b:1;i:56530;b:1;i:56531;b:1;i:56533;b:1;i:56519;b:1;i:56536;b:1;i:56538;b:1;i:56534;i:1;i:56542;b:1;i:56557;b:1;i:56560;b:1;i:56563;b:1;i:56555;i:1;i:56113;i:1;i:56102;i:1;i:56170;i:1;i:56006;i:1;i:56565;i:0;i:56078;i:1;i:56568;i:1;i:56577;b:1;i:56585;b:1;i:56559;b:1;i:56593;b:1;i:56590;b:1;i:56598;b:1;i:56594;b:1;i:56574;b:1;i:56592;b:1;i:56607;i:1;i:56572;i:1;i:56622;i:1;i:56629;b:1;i:56631;i:1;i:56637;i:1;i:56641;i:0;i:56647;i:0;i:56648;i:0;i:56650;i:1;i:56651;i:1;i:56521;i:1;i:56553;i:1;i:56573;i:1;i:56619;i:0;i:56652;i:0;i:56653;i:0;i:56654;i:0;i:55894;i:1;i:56632;b:1;i:56663;b:1;i:56618;i:0;i:56398;i:1;i:56588;i:1;i:56614;i:1;i:56662;i:0;i:56661;i:0;i:56668;b:1;i:56670;b:1;i:56677;b:1;i:56123;i:1;i:56628;b:1;i:56124;i:0;i:56128;i:0;i:56163;i:0;i:56169;i:0;i:56171;i:0;i:56172;i:0;i:56250;i:0;i:56685;b:1;i:56269;i:0;i:56259;i:0;i:56380;i:1;i:56674;i:1;i:56680;i:0;i:56695;b:1;i:56700;b:1;i:56470;b:1;i:56676;b:1;i:56684;b:1;i:56709;b:1;i:56164;i:1;i:56103;i:1;i:56679;i:0;i:56708;i:1;i:56710;i:0;i:56287;i:0;i:56600;i:0;i:56174;i:0;i:56713;b:1;i:56714;i:1;i:56721;i:0;i:56723;i:1;i:56625;b:1;i:56725;i:1;i:56724;i:0;i:56728;b:1;i:56756;b:1;i:56758;b:1;i:56751;i:1;i:56763;b:1;i:56754;b:1;i:56767;b:1;i:56730;b:1;i:56765;b:1;i:56773;b:1;i:56777;i:1;i:56784;i:1;i:56785;i:1;i:56782;i:0;i:56796;b:1;i:56792;i:1;i:56801;b:1;i:56719;b:1;i:56810;b:1;i:56811;b:1;i:56812;b:1;i:56800;b:1;i:56807;b:1;i:56822;i:1;i:56823;b:1;i:56793;b:1;i:56824;b:1;i:56809;b:1;i:56752;b:1;i:56743;b:1;i:56833;b:1;i:56770;b:1;i:56787;b:1;i:56852;b:1;i:56849;i:1;i:56844;i:1;i:56854;b:1;i:56816;b:1;i:56855;b:1;i:56863;b:1;i:56866;b:1;i:56870;b:1;i:56842;b:1;i:56865;b:1;i:56835;b:1;i:56875;b:1;i:56884;i:1;i:56831;i:1;i:56896;b:1;i:56895;b:1;i:56894;b:1;i:56893;b:1;i:56892;b:1;i:56891;b:1;i:56890;b:1;i:56889;b:1;i:56879;b:1;i:56830;i:1;i:56455;i:1;i:56832;i:0;i:56883;i:0;i:56873;i:0;i:56750;i:1;i:56887;i:0;i:56741;i:1;i:56874;i:0;i:56888;i:0;i:56885;i:0;i:56886;i:0;i:56838;i:0;i:56799;i:0;i:56851;i:0;i:56912;b:1;i:56916;b:1;i:56913;i:1;i:56911;i:1;i:56923;i:0;i:56881;b:1;i:56929;i:1;i:56931;b:1;i:56934;i:1;i:56938;b:1;i:56939;b:1;i:56937;i:1;i:56942;i:0;i:56944;b:1;i:56845;b:1;i:56935;b:1;i:56953;b:1;i:56955;b:1;i:56950;i:1;i:56954;b:1;i:56957;b:1;i:56940;b:1;i:56965;b:1;i:56969;b:1;i:56918;b:1;i:56970;b:1;i:56982;i:1;i:56981;i:1;i:56983;b:1;i:56914;b:1;i:56986;b:1;i:56943;b:1;i:56991;b:1;i:56988;b:1;i:56975;b:1;i:56994;b:1;i:56996;b:1;i:56998;b:1;i:57001;b:1;i:57004;b:1;i:57005;i:1;i:56993;i:1;i:56989;i:1;i:56949;i:1;i:57013;b:1;i:57010;b:1;i:57008;i:1;i:57007;i:1;i:56924;b:1;i:57026;b:1;i:57028;b:1;i:57014;i:0;i:57035;b:1;i:57036;b:1;i:57040;b:1;i:57043;i:1;i:57044;i:0;i:57048;i:1;i:57047;i:1;i:57055;i:1;i:57058;i:1;i:57031;b:1;i:57011;b:1;i:57072;b:1;i:57059;b:1;i:57061;i:1;i:57000;b:1;i:57077;b:1;i:57081;b:1;i:57078;i:1;i:57074;i:0;i:57079;i:0;i:57080;i:0;i:57083;i:0;i:57084;i:0;i:57086;b:1;i:57092;i:1;i:57053;b:1;i:57100;b:1;i:57106;b:1;i:57088;b:1;i:57113;b:1;i:57017;b:1;i:57108;i:1;i:57124;b:1;i:57131;b:1;i:57136;b:1;i:57139;b:1;i:57141;b:1;i:57140;b:1;i:57033;b:1;i:57145;b:1;i:57147;b:1;i:57148;b:1;i:57152;b:1;i:57103;b:1;i:57101;b:1;i:57093;b:1;i:57170;b:1;i:57068;b:1;i:57150;i:1;i:57111;i:1;i:57112;i:0;i:57119;i:0;i:57127;i:0;i:57128;i:0;i:57157;i:0;i:57161;i:0;i:57183;i:0;i:57185;i:1;i:57173;b:1;i:57187;i:1;i:57189;i:0;i:57191;i:0;i:57192;i:0;i:57190;i:0;i:57179;i:0;i:57180;i:0;i:57186;i:0;i:57174;b:1;i:57168;b:1;i:57120;i:0;i:57165;b:1;i:57181;i:0;i:57200;b:1;i:57205;b:1;i:57151;b:1;i:57214;b:1;i:57211;i:1;i:57098;i:1;i:57105;i:0;i:57126;i:0;i:57223;b:1;i:57228;i:1;i:57230;b:1;i:57212;b:1;i:57234;b:1;i:55880;i:1;i:57255;b:1;i:57233;b:1;i:57258;b:1;i:57250;i:1;i:57261;b:1;i:57253;i:0;i:57260;i:0;i:57263;b:1;i:57267;b:1;i:57269;b:1;i:57271;b:1;i:57277;b:1;i:57278;b:1;i:57280;b:1;i:57281;b:1;i:57262;i:1;i:57287;b:1;i:57285;b:1;i:57291;b:1;i:57274;i:0;i:57282;i:0;i:57296;b:1;i:57254;i:0;i:57220;b:1;i:57252;b:1;i:57299;i:1;i:57301;i:0;i:57300;i:0;i:57302;i:0;i:57304;i:0;i:57316;b:1;i:57315;b:1;i:57323;b:1;i:57324;b:1;i:57328;b:1;i:57330;b:1;i:57331;b:1;i:57337;b:1;i:57339;b:1;i:57314;b:1;i:57307;b:1;i:57348;b:1;i:57352;b:1;i:57355;b:1;i:57356;b:1;i:57357;b:1;i:57289;b:1;i:57207;b:1;i:57368;b:1;i:57310;i:1;i:57371;i:0;i:57367;i:0;i:57364;i:0;i:57362;i:0;i:57344;b:1;i:57381;b:1;i:57366;b:1;i:57379;b:1;i:57303;i:0;i:57188;i:0;i:57351;i:0;i:57392;b:1;i:57383;b:1;i:57404;b:1;i:57405;b:1;i:57373;b:1;i:57380;b:1;i:57408;b:1;i:57219;b:1;i:57275;b:1;i:57378;b:1;i:57419;b:1;i:57389;i:1;i:57422;i:1;i:57396;i:1;i:57402;i:0;i:57423;i:0;i:57424;i:0;i:57425;i:0;i:57427;i:1;i:57426;i:1;i:57398;i:0;i:57430;b:1;i:57432;b:1;i:57433;b:1;i:57435;b:1;i:57437;i:1;i:57444;b:1;i:57446;b:1;i:57445;i:1;i:57452;b:1;i:57457;b:1;i:57461;b:1;i:57460;b:1;i:57479;b:1;i:57454;i:1;i:57449;i:1;i:57441;b:1;i:57477;i:0;i:57448;i:0;i:57473;i:0;i:57487;b:1;i:56046;b:1;i:57490;b:1;i:57488;b:1;i:57493;b:1;i:57491;b:1;i:57494;i:1;i:57342;b:1;i:57500;b:1;i:57501;i:1;i:57499;i:1;i:57497;i:1;i:57505;i:1;i:57510;b:1;i:57512;i:1;i:57466;b:1;i:57524;b:1;i:57506;b:1;i:57525;i:1;i:57522;i:1;i:57531;b:1;i:57526;b:1;i:57472;b:1;i:57434;b:1;i:57536;b:1;i:57534;b:1;i:57537;i:1;i:57450;b:1;i:57542;b:1;i:57543;b:1;i:57545;b:1;i:57550;b:1;i:57553;b:1;i:57557;b:1;i:57559;i:1;i:57541;b:1;i:57484;b:1;i:57574;i:1;i:57579;b:1;i:57580;b:1;i:57581;b:1;i:57585;b:1;i:57586;b:1;i:57365;b:1;i:57587;i:1;i:57588;i:0;i:57583;i:1;i:57464;i:0;i:57590;b:1;i:57598;b:1;i:57603;i:1;i:57608;b:1;i:57606;i:0;i:57607;i:0;i:57295;b:1;i:57623;b:1;i:57630;b:1;i:57627;i:1;i:57624;i:1;i:57626;i:0;i:57625;i:0;i:57632;b:1;i:57634;i:1;i:57455;i:0;i:57638;i:1;i:57640;i:1;i:57639;i:1;i:57647;b:1;i:57649;b:1;i:57653;b:1;i:57655;b:1;i:57652;b:1;i:57657;i:1;i:57651;i:1;i:57671;b:1;i:57682;b:1;i:57681;b:1;i:57679;i:1;i:57683;i:1;i:57663;i:1;i:57680;b:1;i:57678;b:1;i:57696;i:1;i:57699;i:0;i:57695;i:1;i:57672;i:1;i:57714;b:1;i:57720;b:1;i:57722;b:1;i:54998;b:1;i:57706;b:1;i:57710;b:1;i:57733;i:1;i:57735;b:1;i:57712;b:1;i:57729;i:1;i:57711;i:1;i:57747;i:1;i:57737;i:1;i:57745;i:1;i:57746;i:0;i:57744;i:0;i:57757;i:1;i:57739;b:1;i:57761;b:1;i:57765;b:1;i:57773;b:1;i:57772;b:1;i:57775;b:1;i:57764;i:1;i:57750;i:1;i:57780;i:1;i:57767;b:1;i:57730;b:1;i:57822;b:1;i:57778;b:1;i:57788;i:1;i:57791;i:1;i:57792;i:0;i:57825;b:1;i:57794;i:1;i:57795;i:0;i:57797;i:1;i:57798;i:1;i:57799;i:0;i:57853;b:1;i:57802;i:1;i:57862;b:1;i:57867;b:1;i:57804;i:1;i:57805;i:0;i:57807;i:1;i:57811;i:1;i:57813;b:1;i:57903;b:1;i:57908;b:1;i:57896;i:1;i:57851;i:1;i:57814;i:1;i:57816;i:0;i:57821;i:0;i:57824;i:0;i:57828;i:0;i:57929;b:1;i:57829;i:0;i:57830;i:0;i:57831;i:1;i:57941;b:1;i:57945;b:1;i:57947;b:1;i:57946;b:1;i:57832;i:1;i:57939;b:1;i:57959;b:1;i:57962;b:1;i:57834;i:1;i:57970;b:1;i:57972;b:1;i:57976;b:1;i:57977;b:1;i:57984;b:1;i:57981;b:1;i:57985;b:1;i:57478;b:1;i:57989;i:1;i:57968;i:1;i:57960;i:1;i:57961;i:0;i:57924;i:1;i:57994;b:1;i:57974;b:1;i:58001;b:1;i:58002;b:1;i:58005;b:1;i:57958;b:1;i:58017;b:1;i:58018;b:1;i:58019;b:1;i:58021;b:1;i:58023;b:1;i:57835;i:1;i:57840;i:0;i:57881;i:1;i:57883;i:0;i:57836;i:0;i:57916;i:0;i:57964;i:0;i:57790;i:1;i:58033;i:1;i:58034;b:1;i:58007;b:1;i:58032;i:1;i:57801;i:0;i:57838;i:1;i:58046;b:1;i:58036;b:1;i:58054;b:1;i:58063;i:1;i:58064;i:1;i:58070;b:1;i:58066;b:1;i:58065;b:1;i:58084;b:1;i:58081;b:1;i:58092;i:1;i:58091;i:1;i:57842;i:1;i:58094;i:1;i:57843;i:0;i:57846;i:0;i:58101;i:1;i:58102;b:1;i:58104;b:1;i:58106;b:1;i:58113;b:1;i:58058;b:1;i:58109;i:1;i:58112;b:1;i:58111;b:1;i:58105;b:1;i:58117;i:1;i:58030;i:1;i:58107;b:1;i:58133;b:1;i:58132;i:1;i:58127;i:1;i:58126;i:1;i:58118;i:1;i:57892;i:1;i:58146;b:1;i:58145;b:1;i:57906;i:1;i:57923;i:1;i:57965;i:1;i:57966;i:0;i:58022;i:0;i:58086;b:1;i:58167;b:1;i:58171;b:1;i:58180;b:1;i:58173;b:1;i:58182;b:1;i:57983;b:1;i:58157;i:1;i:58158;i:1;i:58160;i:1;i:58162;i:1;i:58163;i:0;i:58165;i:0;i:58166;i:0;i:58170;i:0;i:58177;i:0;i:58187;i:0;i:58188;i:0;i:58193;i:0;i:58197;i:0;i:58198;i:0;i:58096;i:1;i:58097;i:1;i:58098;i:1;i:58125;i:0;i:58124;i:1;i:58138;i:0;i:58139;i:0;i:58140;i:0;i:58141;i:0;i:58144;i:0;i:58143;i:0;i:58151;i:0;i:58152;i:0;i:58156;i:0;i:58169;i:0;i:57953;i:0;i:58199;i:1;i:58042;i:0;i:58052;i:0;i:58062;i:0;i:58088;i:0;i:58205;b:1;i:58128;i:1;i:58129;i:0;i:58130;i:0;i:58136;i:1;i:58148;i:0;i:58202;i:1;i:58201;i:1;i:58194;i:0;i:58192;i:0;i:58175;i:0;i:58012;i:1;i:57882;i:0;i:57922;i:0;i:58040;i:1;i:58089;i:0;i:58119;i:0;i:58217;b:1;i:58219;b:1;i:58221;b:1;i:58218;b:1;i:57833;i:1;i:58232;b:1;i:58228;b:1;i:58220;b:1;i:58238;b:1;i:58227;b:1;i:58242;b:1;i:58247;b:1;i:58229;i:1;i:58230;i:1;i:58231;i:1;i:58240;i:1;i:58243;i:1;i:58244;i:1;i:58245;i:1;i:58262;i:1;i:58256;b:1;i:58233;i:0;i:58251;i:0;i:58254;i:0;i:58261;i:0;i:58259;i:0;i:58257;i:0;i:58263;i:1;i:58264;i:0;i:58314;b:1;i:58315;b:1;i:58316;b:1;i:58323;b:1;i:58258;b:1;i:58359;i:0;i:58322;b:1;i:58365;b:1;i:58351;b:1;i:58364;b:1;i:58252;b:1;i:58330;b:1;i:58331;b:1;i:58384;b:1;i:58393;b:1;i:55550;i:1;i:55685;i:1;i:55349;i:1;i:58260;i:1;i:23618;i:1;i:58305;b:1;i:58404;b:1;i:58407;b:1;i:12224;i:1;i:23855;i:1;i:24734;i:1;i:24636;i:1;i:24831;i:1;i:25089;i:1;i:25688;i:1;i:25852;i:1;i:26334;i:1;i:26394;i:1;i:27018;i:1;i:33082;i:1;i:33384;i:1;i:33965;i:1;i:37401;i:1;i:46410;i:1;i:38721;i:1;i:48860;i:1;i:49189;i:1;i:51147;i:1;i:52377;i:1;i:58431;b:1;i:58443;b:1;i:58442;b:1;i:58324;b:1;i:58448;b:1;i:58449;b:1;i:58458;b:1;i:58461;b:1;i:58476;i:1;i:58483;b:1;i:58496;b:1;i:58508;b:1;i:58472;b:1;i:58510;b:1;i:58507;b:1;i:58511;b:1;i:58504;b:1;i:58467;i:1;i:58396;i:1;i:58519;b:1;i:58378;i:0;i:58269;i:0;i:58518;i:0;i:58517;i:0;i:58491;i:0;i:58490;i:0;i:58392;i:1;i:58494;i:0;i:58502;b:1;i:58534;b:1;i:58505;b:1;i:58474;b:1;i:58403;b:1;i:58540;b:1;i:58543;b:1;i:58549;b:1;i:58554;b:1;i:58557;b:1;i:58550;b:1;i:58538;b:1;i:58552;b:1;i:57975;b:1;i:58497;i:0;i:58570;b:1;i:58539;i:1;i:58566;i:0;i:58571;i:0;i:58471;i:0;i:58575;b:1;i:58578;b:1;i:58580;b:1;i:58585;b:1;i:58584;b:1;i:58568;b:1;i:58594;b:1;i:58603;b:1;i:58605;b:1;i:58473;b:1;i:58672;b:1;i:58625;i:1;i:58591;i:1;i:58574;i:0;i:58682;b:1;i:58685;b:1;i:58416;i:1;i:58203;i:1;i:58209;i:1;i:58668;i:0;i:58681;i:0;i:58701;b:1;i:58703;b:1;i:58705;b:1;i:58688;b:1;i:58706;b:1;i:58711;b:1;i:58717;b:1;i:58677;b:1;i:58656;i:1;i:58729;b:1;i:16355;i:1;i:52314;i:1;i:38519;i:1;i:27644;i:1;i:25471;i:1;i:33724;i:1;i:43352;i:0;i:27123;i:1;i:35990;i:1;i:52288;i:1;i:35823;i:1;i:55507;i:1;i:33528;i:1;i:32656;i:1;i:24114;i:1;i:34607;i:1;i:54277;i:1;i:38770;i:1;i:47957;i:0;i:57925;i:1;i:30322;i:1;i:20846;i:1;i:58738;b:1;i:53259;i:1;i:34779;i:1;i:52615;i:1;i:45258;i:1;i:52637;i:1;i:51928;i:1;i:52982;i:0;i:52331;i:0;i:58741;b:1;i:58742;b:1;i:58737;b:1;i:58746;b:1;i:58745;b:1;i:45041;i:1;i:48467;i:1;i:21670;i:1;i:38167;i:1;i:34600;i:1;i:24395;i:1;i:38320;i:1;i:38895;i:1;i:36021;i:1;i:58215;i:1;i:58732;i:0;i:58728;i:0;i:58708;i:0;i:58667;i:0;i:35306;i:1;i:58752;b:1;i:58751;b:1;i:15236;i:1;i:16709;i:1;i:58761;b:1;i:58762;b:1;i:57789;i:1;i:57969;i:0;i:58003;i:0;i:58053;i:0;i:58270;i:0;i:58272;i:0;i:58276;i:0;i:58280;i:0;i:58281;i:0;i:58286;i:0;i:58287;i:0;i:58288;i:0;i:58291;i:0;i:58299;i:0;i:58300;i:0;i:58301;i:0;i:58304;i:0;i:57839;i:0;i:58341;i:0;i:57879;i:0;i:55949;i:1;i:58358;i:0;i:58361;i:0;i:58362;i:0;i:58370;i:0;i:58371;i:0;i:58379;i:0;i:58391;i:0;i:58399;i:0;i:58412;i:0;i:58455;i:0;i:58480;i:0;i:58501;i:0;i:58513;i:0;i:58602;i:0;i:58607;i:0;i:58609;i:0;i:58611;i:0;i:58612;i:0;i:58615;i:0;i:58618;i:0;i:58622;i:0;i:58629;i:0;i:58630;i:0;i:58636;i:0;i:55589;i:1;i:58643;i:0;i:58644;i:0;i:58649;i:0;i:58650;i:0;i:55736;i:1;i:58652;i:0;i:58654;i:0;i:58657;i:0;i:58661;i:0;i:58664;i:0;i:58670;i:0;i:58690;i:0;i:58696;i:0;i:58802;b:1;i:53326;i:1;i:58675;i:0;i:58673;i:0;i:58620;i:0;i:58610;i:0;i:58369;i:0;i:58292;i:0;i:55793;i:1;i:57796;i:0;i:57850;i:0;i:57893;i:0;i:57914;i:0;i:57926;i:0;i:57951;i:0;i:58767;i:0;i:58613;i:0;i:58310;i:0;i:55772;i:1;i:57866;i:0;i:57868;i:0;i:57869;i:0;i:58271;i:0;i:58278;i:0;i:58360;i:0;i:58614;i:0;i:58632;i:0;i:58646;i:0;i:58694;i:0;i:58702;i:0;i:58294;i:0;i:58820;b:1;i:58827;b:1;i:58831;b:1;i:58822;b:1;i:58836;b:1;i:58840;b:1;i:58800;b:1;i:55813;i:1;i:58274;i:1;i:58846;b:1;i:55888;i:1;i:55658;i:1;i:57863;i:0;i:58103;i:0;i:48413;i:1;i:58856;i:1;i:17277;i:0;i:19944;i:0;i:58881;b:1;i:32092;i:1;i:58885;b:1;i:58865;b:1;i:25622;i:1;i:58889;b:1;i:58897;b:1;i:58894;b:1;i:58896;b:1;i:58901;b:1;i:58903;b:1;i:58900;b:1;i:58824;b:1;i:58899;b:1;i:58859;b:1;i:11994;i:1;i:58826;i:0;i:58839;i:0;i:34974;i:1;i:58598;i:0;i:58601;i:0;i:58621;i:0;i:58631;i:0;i:58913;i:1;i:58409;i:0;i:58195;i:0;i:58417;i:0;i:58303;i:0;i:58422;i:0;i:58226;i:0;i:58427;i:0;i:58915;b:1;i:58524;i:0;i:58562;i:0;i:58693;i:0;i:58676;i:0;i:58697;i:0;i:58768;i:0;i:58825;i:0;i:58855;i:0;i:58858;i:0;i:58871;i:0;i:58872;i:0;i:58876;i:0;i:58880;i:0;i:58920;b:1;i:58934;i:0;i:58919;i:1;i:58939;b:1;i:58918;i:1;i:58927;b:1;i:58949;b:1;i:58861;b:1;i:58863;b:1;i:58904;i:0;i:58847;i:0;i:58961;b:1;i:58733;b:1;i:58981;b:1;i:58986;b:1;i:58987;b:1;i:58989;b:1;i:58990;b:1;i:58993;b:1;i:58849;b:1;i:58996;b:1;i:58967;b:1;i:58994;b:1;i:58995;b:1;i:58887;b:1;i:58755;b:1;i:59002;b:1;i:59003;b:1;i:59000;b:1;i:59010;b:1;i:58726;b:1;i:57904;b:1;i:58725;b:1;i:58731;b:1;i:58730;b:1;i:57901;b:1;i:58965;b:1;i:58980;i:1;i:58979;i:1;i:59053;b:1;i:59031;b:1;i:59058;b:1;i:59081;b:1;i:59082;b:1;i:59094;b:1;i:59103;b:1;i:59091;i:0;i:59061;i:0;i:59076;i:0;i:59006;i:0;i:59108;b:1;i:59118;b:1;i:59102;b:1;i:59041;b:1;i:59143;b:1;i:59112;b:1;i:59131;i:0;i:59150;b:1;i:59161;b:1;i:59162;b:1;i:59170;b:1;i:59176;b:1;i:59174;b:1;i:59008;i:1;i:59177;b:1;i:59183;b:1;i:59187;b:1;i:59092;b:1;i:59080;b:1;i:59192;i:1;i:58684;b:1;i:59205;b:1;i:59207;b:1;i:59038;b:1;i:58966;b:1;i:59197;b:1;i:59075;b:1;i:59074;b:1;i:59072;b:1;i:59071;b:1;i:59069;b:1;i:59068;b:1;i:59067;b:1;i:59066;b:1;i:59065;b:1;i:59064;b:1;i:59063;b:1;i:59059;b:1;i:59055;b:1;i:59049;b:1;i:59048;b:1;i:59046;b:1;i:59045;b:1;i:59044;b:1;i:59043;b:1;i:59042;b:1;i:59040;b:1;i:59028;b:1;i:59027;b:1;i:57897;b:1;i:59026;b:1;i:58968;b:1;i:58727;b:1;i:57913;b:1;i:57912;b:1;i:57905;b:1;i:57910;b:1;i:57911;b:1;i:59039;b:1;i:59219;i:0;i:59152;b:1;i:59171;b:1;i:59211;b:1;i:59239;b:1;i:59242;b:1;i:59070;b:1;i:59062;b:1;i:59142;b:1;i:59047;b:1;i:59233;i:0;i:59226;i:0;i:59252;b:1;i:59253;b:1;i:59257;b:1;i:41431;i:1;i:59256;i:1;i:59263;b:1;i:59078;b:1;i:59268;b:1;i:59208;b:1;i:59269;b:1;i:59270;b:1;i:59271;b:1;i:58743;b:1;i:59188;b:1;i:59203;b:1;i:59273;b:1;i:59274;b:1;i:59276;b:1;i:59191;b:1;i:59262;b:1;i:59247;b:1;i:59279;i:1;i:59290;b:1;i:59295;b:1;i:59241;b:1;i:59238;b:1;i:59275;b:1;i:58942;b:1;i:59326;b:1;i:59167;b:1;i:59280;i:1;i:59311;i:0;i:59320;i:0;i:59329;i:0;i:59116;b:1;i:59333;b:1;i:59336;b:1;i:59301;b:1;i:59342;b:1;i:59304;b:1;i:59234;b:1;i:59299;b:1;i:59193;b:1;i:59352;b:1;i:59355;b:1;i:59364;b:1;i:59370;b:1;i:59378;b:1;i:59368;b:1;i:59050;b:1;i:59384;b:1;i:59373;i:1;i:59390;b:1;i:59393;b:1;i:59400;b:1;i:59401;b:1;i:59403;b:1;i:59407;b:1;i:59399;b:1;i:59372;b:1;i:59420;b:1;i:59422;b:1;i:59289;i:1;i:59431;i:0;i:59402;i:0;i:59440;b:1;i:59382;i:0;i:59432;i:0;i:59445;b:1;i:59459;i:1;i:59450;i:1;i:59454;i:1;i:59340;i:1;i:59472;i:1;i:59486;b:1;i:59478;i:1;i:59497;b:1;i:59254;b:1;i:59416;b:1;i:59499;b:1;i:59377;b:1;i:59479;b:1;i:59505;b:1;i:59397;b:1;i:59500;b:1;i:59419;b:1;i:59495;b:1;i:59509;b:1;i:59501;b:1;i:59512;b:1;i:59519;b:1;i:59521;b:1;i:59529;i:1;i:59513;i:1;i:59493;i:1;i:59524;i:1;i:59533;i:0;i:59475;i:1;i:59527;b:1;i:59538;b:1;i:59543;b:1;i:59545;b:1;i:59534;b:1;i:59549;i:1;i:59546;b:1;i:59547;b:1;i:59556;b:1;i:59560;b:1;i:59541;b:1;i:59568;b:1;i:59577;b:1;i:59584;b:1;i:59423;b:1;i:59539;b:1;i:59515;b:1;i:59592;b:1;i:59571;b:1;i:59434;b:1;i:59460;b:1;i:59611;b:1;i:59606;i:0;i:59578;i:0;i:59613;i:1;i:59462;b:1;i:59625;b:1;i:58608;i:1;i:59627;i:1;i:59628;i:1;i:59635;i:0;i:59619;i:1;i:59621;i:1;i:59620;i:1;i:59622;i:1;i:59643;i:1;i:59645;b:1;i:59648;b:1;i:59639;b:1;i:59644;b:1;i:59650;b:1;i:59649;b:1;i:59661;b:1;i:59629;b:1;i:59647;i:1;i:59656;i:0;i:59669;b:1;i:59667;b:1;i:59659;i:1;i:59676;b:1;i:59678;i:1;i:59685;b:1;i:59660;b:1;i:59686;b:1;i:59687;b:1;i:59690;b:1;i:59693;b:1;i:59701;b:1;i:59703;b:1;i:59705;b:1;i:59706;b:1;i:59709;b:1;i:59717;b:1;i:59721;b:1;i:59710;b:1;i:59707;i:1;i:59200;b:1;i:59636;b:1;i:59741;i:1;i:59684;b:1;i:59765;b:1;i:59713;b:1;i:59772;b:1;i:59744;b:1;i:59738;b:1;i:59787;b:1;i:59789;b:1;i:59786;b:1;i:59795;b:1;i:59788;b:1;i:59791;i:0;i:59799;b:1;i:59779;i:0;i:59780;i:0;i:59797;b:1;i:59796;b:1;i:59814;b:1;i:59811;b:1;i:59821;b:1;i:59832;b:1;i:59849;b:1;i:59804;b:1;i:59856;b:1;i:59715;b:1;i:59855;b:1;i:59860;b:1;i:59861;b:1;i:59700;b:1;i:59857;b:1;i:59865;b:1;i:59761;b:1;i:59872;b:1;i:59848;i:0;i:59841;b:1;i:59884;b:1;i:59874;i:0;i:59880;b:1;i:59565;b:1;i:59916;b:1;i:59923;b:1;i:59927;b:1;i:59928;b:1;i:59822;b:1;i:59956;b:1;i:59891;i:1;i:59910;i:0;i:59919;i:0;i:59920;i:0;i:59948;i:1;i:59966;b:1;i:59967;b:1;i:59969;b:1;i:59970;i:1;i:59980;b:1;i:59937;b:1;i:59899;b:1;i:59987;b:1;i:59992;b:1;i:59996;b:1;i:59853;b:1;i:59863;i:0;i:59978;i:1;i:59986;i:0;i:60008;i:0;i:60018;b:1;i:59924;b:1;i:60028;b:1;i:60029;b:1;i:60031;b:1;i:60040;b:1;i:60045;b:1;i:60048;b:1;i:60051;b:1;i:60027;b:1;i:60046;b:1;i:60061;b:1;i:60068;b:1;i:60020;b:1;i:60094;b:1;i:58660;b:1;i:60107;b:1;i:60108;b:1;i:60111;b:1;i:60112;b:1;i:60113;b:1;i:60114;b:1;i:60072;b:1;i:60121;i:0;i:59905;i:1;i:60129;i:0;i:60128;i:0;i:60127;i:0;i:60096;b:1;i:60099;b:1;i:60133;b:1;i:60140;b:1;i:60150;b:1;i:60151;b:1;i:60152;b:1;i:60157;b:1;i:60073;b:1;i:60034;b:1;i:60143;b:1;i:60136;i:1;i:60168;b:1;i:60110;b:1;i:60173;b:1;i:60175;b:1;i:60179;b:1;i:60183;b:1;i:60188;b:1;i:60180;b:1;i:60164;b:1;i:60177;b:1;i:60178;i:1;i:60156;b:1;i:60202;b:1;i:60208;b:1;i:60212;b:1;i:60207;b:1;i:60197;i:0;i:60198;i:0;i:60201;i:0;i:60215;i:0;i:60224;b:1;i:60227;b:1;i:60176;b:1;i:60229;b:1;i:60149;b:1;i:60232;b:1;i:60210;b:1;i:60231;b:1;i:60228;b:1;i:60238;b:1;i:60242;b:1;i:60247;b:1;i:60248;i:0;i:60249;b:1;i:60251;b:1;i:60257;b:1;i:60266;b:1;i:60270;b:1;i:60271;b:1;i:60292;b:1;i:60288;i:1;i:60276;b:1;i:60279;b:1;i:60261;b:1;i:60298;b:1;i:60277;b:1;i:60293;b:1;i:60309;b:1;i:60310;b:1;i:60301;b:1;i:60318;b:1;i:60319;b:1;i:60317;b:1;i:60320;b:1;i:60330;b:1;i:60334;b:1;i:60306;i:1;i:60329;i:0;i:60326;i:0;i:60322;i:0;i:60339;b:1;i:60344;b:1;i:60347;b:1;i:60358;b:1;i:60364;b:1;i:60336;b:1;i:60348;b:1;i:60323;b:1;i:60367;b:1;i:60307;b:1;i:60305;i:1;i:60379;b:1;i:60388;b:1;i:60387;b:1;i:60390;b:1;i:60393;b:1;i:60400;b:1;i:60403;b:1;i:60408;b:1;i:60399;b:1;i:60413;b:1;i:60414;b:1;i:60415;b:1;i:60421;b:1;i:60423;b:1;i:60395;b:1;i:60428;b:1;i:60432;b:1;i:60426;b:1;i:60375;b:1;i:60418;i:1;i:60440;b:1;i:60442;b:1;i:60315;b:1;i:60448;b:1;i:60431;b:1;i:60451;b:1;i:60297;b:1;i:60424;b:1;i:60462;b:1;i:60461;b:1;i:60419;b:1;i:60456;b:1;i:60365;b:1;i:60447;b:1;i:60386;b:1;i:60496;b:1;i:60498;i:1;i:60491;b:1;i:60482;b:1;i:60495;b:1;i:60446;b:1;i:60488;i:0;i:60193;b:1;i:60511;b:1;i:60499;i:0;i:60503;i:1;i:60515;b:1;i:60389;b:1;i:60500;b:1;i:60524;b:1;i:60508;i:1;i:60478;i:0;i:60516;i:0;i:60517;i:0;i:60526;i:0;i:60523;b:1;i:60514;b:1;i:60545;b:1;i:60460;b:1;i:60549;b:1;i:60556;b:1;i:60539;b:1;i:60560;b:1;i:60534;i:0;i:60580;b:1;i:60552;b:1;i:60555;b:1;i:60569;i:0;i:60581;i:0;i:60340;b:1;i:60542;b:1;i:60577;b:1;i:60602;b:1;i:60604;b:1;i:60611;b:1;i:60570;b:1;i:60635;b:1;i:60641;b:1;i:60644;b:1;i:60643;b:1;i:60665;b:1;i:60664;b:1;i:60615;b:1;i:60667;b:1;i:60598;i:0;i:60626;i:0;i:60640;b:1;i:60662;i:0;i:60650;i:0;i:60627;i:0;i:60633;i:0;i:60596;i:1;i:60672;b:1;i:60679;b:1;i:60684;b:1;i:60659;b:1;i:60685;b:1;i:60691;b:1;i:40660;b:1;i:60692;i:0;i:60646;b:1;i:60704;b:1;i:60694;b:1;i:60601;b:1;i:60703;i:1;i:60638;b:1;i:60700;i:1;i:60708;b:1;i:60710;b:1;i:60649;b:1;i:60670;b:1;i:60715;b:1;i:60728;b:1;i:60727;b:1;i:60733;b:1;i:60736;b:1;i:60755;b:1;i:60756;b:1;i:60745;b:1;i:60758;b:1;i:60687;b:1;i:60770;b:1;i:60763;b:1;i:60768;b:1;i:60772;b:1;i:60776;b:1;i:60778;b:1;i:60779;b:1;i:60752;b:1;i:60766;i:1;i:60791;b:1;i:60797;b:1;i:60384;b:1;i:60735;b:1;i:60799;b:1;i:60803;b:1;i:60810;b:1;i:60565;b:1;i:60811;b:1;i:60827;b:1;i:60830;b:1;i:60822;i:1;i:60814;b:1;i:60832;i:1;i:60839;b:1;i:60837;b:1;i:60840;b:1;i:60843;b:1;i:60842;b:1;i:60824;b:1;i:60853;b:1;i:60863;b:1;i:60868;b:1;i:60875;b:1;i:60858;b:1;i:60876;b:1;i:60882;b:1;i:60818;i:1;i:60879;i:0;i:60878;i:0;i:60874;i:0;i:60794;b:1;i:60884;b:1;i:60883;b:1;i:60880;i:0;i:60877;i:0;i:60889;b:1;i:60890;b:1;i:60893;b:1;i:60900;b:1;i:60903;b:1;i:60904;b:1;i:60902;b:1;i:60901;b:1;i:60899;b:1;i:60918;b:1;i:60919;b:1;i:60917;i:0;i:60923;b:1;i:60881;b:1;i:60933;b:1;i:60936;b:1;i:60938;b:1;i:60944;b:1;i:54526;i:1;i:60930;i:1;i:60942;i:0;i:60946;b:1;i:60934;b:1;i:60954;b:1;i:60956;b:1;i:60958;b:1;i:60947;b:1;i:60957;i:1;i:60951;i:1;i:60965;i:1;i:60885;b:1;i:60519;b:1;i:60975;i:1;i:60977;i:0;i:60979;b:1;i:60982;b:1;i:60972;b:1;i:60987;b:1;i:60897;b:1;i:60989;b:1;i:60984;b:1;i:60980;i:1;i:60986;i:0;i:60991;i:0;i:60992;i:0;i:60978;b:1;i:61002;b:1;i:60994;i:1;i:61006;i:0;i:60983;b:1;i:61015;b:1;i:61014;b:1;i:61022;b:1;i:61036;b:1;i:61038;b:1;i:61018;i:1;i:61023;i:0;i:61027;i:0;i:61028;i:0;i:61025;i:0;i:61040;b:1;i:60739;b:1;i:61042;b:1;i:61043;b:1;i:61031;b:1;i:61029;b:1;i:61046;b:1;i:60988;b:1;i:61035;b:1;i:61055;b:1;i:61058;b:1;i:61064;b:1;i:61065;b:1;i:61066;b:1;i:61057;b:1;i:61067;b:1;i:61053;i:1;i:61073;b:1;i:61068;b:1;i:61077;b:1;i:61081;b:1;i:61087;b:1;i:61089;b:1;i:61071;b:1;i:61082;b:1;i:61091;b:1;i:61079;b:1;i:61019;b:1;i:61095;b:1;i:61094;b:1;i:61100;b:1;i:61099;b:1;i:61102;i:1;i:61101;i:1;i:61076;b:1;i:61109;b:1;i:61108;b:1;i:61051;b:1;i:61116;b:1;i:61118;b:1;i:61111;i:1;i:61112;i:1;i:61120;b:1;i:61104;b:1;i:61088;b:1;i:61119;i:1;i:61124;b:1;i:61126;b:1;i:61127;b:1;i:61113;b:1;i:61129;b:1;i:61135;b:1;i:61138;b:1;i:61125;i:1;i:61153;b:1;i:61151;b:1;i:61140;b:1;i:61158;b:1;i:61106;b:1;i:61159;b:1;i:61011;b:1;i:61156;b:1;i:61177;b:1;i:61182;b:1;i:60721;b:1;i:60709;b:1;i:61157;b:1;i:61198;b:1;i:61200;b:1;i:61178;i:1;i:61196;i:1;i:61201;b:1;i:61169;i:1;i:61170;i:0;i:61167;b:1;i:61165;b:1;i:61220;b:1;i:61176;b:1;i:61214;i:1;i:61225;b:1;i:61215;i:0;i:61228;b:1;i:61229;b:1;i:61234;b:1;i:61236;b:1;i:61249;b:1;i:61242;b:1;i:61240;b:1;i:61233;b:1;i:61230;b:1;i:61254;b:1;i:61255;b:1;i:61245;b:1;i:61251;b:1;i:61248;i:1;i:61284;b:1;i:61291;i:1;i:61279;i:1;i:61293;b:1;i:61047;b:1;i:61296;b:1;i:61300;b:1;i:61324;b:1;i:61325;i:1;i:61336;b:1;i:61334;b:1;i:61331;b:1;i:61333;b:1;i:61190;b:1;i:61357;b:1;i:61358;b:1;i:61365;i:1;i:61289;b:1;i:61369;b:1;i:61376;b:1;i:61380;b:1;i:61335;b:1;i:61382;b:1;i:61383;b:1;i:61393;i:1;i:61400;i:0;i:61363;b:1;i:61386;b:1;i:61398;b:1;i:61403;i:1;i:61409;i:0;i:61423;b:1;i:61430;b:1;i:61435;b:1;i:61436;i:1;i:61438;b:1;i:61437;b:1;i:61411;b:1;i:61394;b:1;i:61442;b:1;i:61319;b:1;i:61218;b:1;i:61428;b:1;i:61458;b:1;i:61451;b:1;i:61454;b:1;i:61453;b:1;i:61466;b:1;i:61473;b:1;i:61474;b:1;i:61481;b:1;i:61486;b:1;i:61487;b:1;i:61469;b:1;i:61477;i:1;i:61498;b:1;i:61478;b:1;i:61426;b:1;i:61508;b:1;i:61507;b:1;i:61483;b:1;i:61460;b:1;i:61521;b:1;i:61505;i:1;i:61524;i:0;i:61464;b:1;i:61535;b:1;i:61539;i:1;i:61537;i:1;i:61449;b:1;i:61542;b:1;i:61545;b:1;i:61551;b:1;i:61407;b:1;i:61506;b:1;i:61546;i:1;i:61511;b:1;i:61548;b:1;i:61566;i:1;i:61560;b:1;i:61571;b:1;i:61485;b:1;i:61579;b:1;i:61600;b:1;i:61594;b:1;i:61616;b:1;i:61612;b:1;i:61606;i:0;i:61623;b:1;i:61625;b:1;i:61573;b:1;i:61588;b:1;i:61568;b:1;i:61572;b:1;i:61617;b:1;i:61613;b:1;i:61631;b:1;i:61628;b:1;i:61632;b:1;i:61636;i:1;i:61637;b:1;i:61639;b:1;i:61599;b:1;i:61626;i:1;i:61627;b:1;i:61648;b:1;i:61622;b:1;i:61651;b:1;i:61650;b:1;i:61610;b:1;i:61658;b:1;i:61656;i:1;i:61666;b:1;i:61668;b:1;i:61665;b:1;i:61581;b:1;i:61657;b:1;i:61518;b:1;i:61674;b:1;i:61677;b:1;i:61660;b:1;i:61620;b:1;i:61700;b:1;i:61709;b:1;i:61711;b:1;i:61634;b:1;i:61713;b:1;i:61684;b:1;i:61729;b:1;i:61733;b:1;i:61712;b:1;i:61649;b:1;i:61716;b:1;i:61736;b:1;i:61740;b:1;i:61728;b:1;i:61721;b:1;i:60302;b:1;i:61750;b:1;i:61757;b:1;i:61678;i:1;i:61679;i:0;i:61766;b:1;i:61779;b:1;i:61780;b:1;i:61763;b:1;i:61784;b:1;i:61787;b:1;i:61774;b:1;i:61804;b:1;i:61803;b:1;i:61708;i:1;i:61810;b:1;i:61725;i:0;i:61795;i:1;i:61812;i:0;i:61813;i:0;i:61796;i:0;i:61821;b:1;i:61824;b:1;i:61715;i:0;i:61714;i:0;i:61690;i:0;i:61687;i:0;i:61828;b:1;i:61827;b:1;i:61829;b:1;i:61831;b:1;i:61834;b:1;i:61835;b:1;i:61846;b:1;i:61847;b:1;i:61848;b:1;i:61832;b:1;i:61844;b:1;i:61842;i:1;i:61849;b:1;i:61809;b:1;i:61820;b:1;i:61850;b:1;i:61858;b:1;i:61860;b:1;i:61870;b:1;i:61865;i:1;i:61859;i:1;i:61863;i:0;i:61864;i:0;i:61869;i:0;i:61861;b:1;i:61880;b:1;i:61851;b:1;i:61888;b:1;i:61892;b:1;i:61874;b:1;i:61895;b:1;i:61881;b:1;i:61898;b:1;i:61899;b:1;i:61902;b:1;i:61889;i:1;i:61904;i:0;i:61908;i:0;i:61905;i:0;i:61906;i:0;i:61907;i:0;i:61910;b:1;i:61909;b:1;i:61896;b:1;i:61912;i:1;i:61925;b:1;i:61921;b:1;i:61933;b:1;i:61930;b:1;i:61932;b:1;i:61950;b:1;i:61924;b:1;i:61951;b:1;i:61953;b:1;i:61955;b:1;i:61943;i:1;i:61944;i:0;i:61949;i:0;i:61952;i:0;i:61856;b:1;i:61928;i:1;i:61963;b:1;i:61961;i:1;i:61965;i:0;i:61969;b:1;i:61973;b:1;i:61979;b:1;i:61983;b:1;i:61958;b:1;i:61987;b:1;i:61989;b:1;i:61974;i:1;i:61988;i:0;i:61984;i:0;i:61990;b:1;i:62002;b:1;i:62006;b:1;i:62007;b:1;i:62008;b:1;i:62009;b:1;i:62010;b:1;i:62012;b:1;i:62013;b:1;i:62018;i:1;i:62023;b:1;i:62022;b:1;i:61997;i:1;i:61998;i:0;i:62001;i:0;i:62016;i:0;i:62036;b:1;i:62037;b:1;i:62031;i:1;i:62047;b:1;i:62049;b:1;i:62050;b:1;i:62051;b:1;i:62025;b:1;i:62054;b:1;i:62059;b:1;i:62043;b:1;i:62033;b:1;i:62063;b:1;i:62064;b:1;i:62071;b:1;i:62085;b:1;i:62087;b:1;i:62088;i:1;i:62102;b:1;i:62106;b:1;i:62109;b:1;i:62104;b:1;i:62113;b:1;i:62100;b:1;i:62120;b:1;i:62127;b:1;i:62130;b:1;i:61817;b:1;i:62139;b:1;i:62115;b:1;i:62112;b:1;i:62144;b:1;i:62150;b:1;i:62149;b:1;i:62146;b:1;i:62153;b:1;i:62151;b:1;i:62156;b:1;i:62158;b:1;i:62160;b:1;i:62162;b:1;i:62164;b:1;i:62165;i:1;i:62126;i:1;i:62185;b:1;i:62183;i:1;i:62184;i:0;i:62188;i:0;i:62189;b:1;i:62166;i:1;i:62192;i:0;i:62191;i:1;i:62178;b:1;i:62198;i:0;i:62215;i:0;i:62216;i:0;i:62217;i:0;i:62218;i:0;i:62213;i:0;i:62212;i:0;i:62221;b:1;i:62223;b:1;i:62201;b:1;i:62220;i:1;i:62206;b:1;i:62181;b:1;i:62234;i:1;i:62238;b:1;i:62239;b:1;i:62240;b:1;i:62243;b:1;i:62228;i:1;i:62235;i:0;i:62245;i:0;i:62249;b:1;i:62122;b:1;i:62256;b:1;i:62230;i:1;i:62264;b:1;i:62276;b:1;i:62279;b:1;i:62278;b:1;i:62281;i:1;i:62283;b:1;i:62284;b:1;i:62259;i:0;i:62258;i:1;i:62246;b:1;i:62295;b:1;i:62294;i:1;i:62293;i:1;i:62291;b:1;i:62304;b:1;i:62306;b:1;i:62316;b:1;i:62296;b:1;i:62320;i:0;i:62303;i:0;i:62313;b:1;i:62327;b:1;i:62328;b:1;i:62331;b:1;i:62310;b:1;i:62340;b:1;i:62335;i:1;i:62330;b:1;i:62332;b:1;i:62299;b:1;i:62348;b:1;i:62337;i:1;i:62354;i:0;i:62353;i:0;i:62338;i:0;i:62352;i:1;i:62205;b:1;i:62356;b:1;i:62358;b:1;i:62364;b:1;i:62377;b:1;i:62378;b:1;i:62379;b:1;i:62355;b:1;i:62385;b:1;i:62315;b:1;i:62366;i:1;i:62367;i:0;i:62368;i:0;i:62369;i:0;i:62375;i:0;i:62389;i:0;i:62391;b:1;i:62262;b:1;i:62393;i:1;i:62395;b:1;i:62363;b:1;i:62406;b:1;i:62227;b:1;i:62376;b:1;i:62419;b:1;i:62415;b:1;i:62388;b:1;i:62422;b:1;i:62425;b:1;i:62387;b:1;i:62435;b:1;i:62437;b:1;i:62396;b:1;i:62439;b:1;i:62441;b:1;i:62447;b:1;i:62444;b:1;i:62455;b:1;i:62474;b:1;i:62466;b:1;i:62484;b:1;i:62486;b:1;i:62489;b:1;i:62495;b:1;i:62470;b:1;i:62504;b:1;i:62420;i:1;i:62426;i:0;i:62440;i:0;i:62506;b:1;i:62507;b:1;i:62511;b:1;i:62514;b:1;i:62513;b:1;i:62493;i:0;i:62465;i:1;i:62516;b:1;i:62448;i:0;i:62468;b:1;i:62523;b:1;i:62488;i:1;i:62411;i:1;i:62402;i:1;i:62491;b:1;i:62542;b:1;i:62530;i:1;i:62538;i:0;i:62543;b:1;i:62345;b:1;i:62547;i:1;i:62548;i:0;i:62551;b:1;i:62549;b:1;i:62540;b:1;i:62556;b:1;i:62558;b:1;i:62561;b:1;i:62565;b:1;i:62570;b:1;i:62572;b:1;i:62579;b:1;i:62581;b:1;i:62582;b:1;i:62583;i:1;i:62569;i:1;i:62559;i:1;i:62560;i:1;i:62589;b:1;i:62591;b:1;i:62595;b:1;i:62599;b:1;i:62602;b:1;i:62587;i:1;i:62603;i:0;i:62610;b:1;i:62615;b:1;i:62616;b:1;i:62618;b:1;i:62626;b:1;i:62621;i:1;i:62632;i:1;i:62633;i:1;i:62636;b:1;i:62641;b:1;i:62617;b:1;i:62643;b:1;i:62646;b:1;i:62648;b:1;i:62594;b:1;i:62652;b:1;i:62660;b:1;i:62662;b:1;i:62651;b:1;i:62664;b:1;i:62627;b:1;i:62666;i:1;i:62672;i:1;i:62674;i:1;i:62675;i:0;i:62553;b:1;i:62550;b:1;i:62667;b:1;i:62685;b:1;i:62686;b:1;i:62695;i:1;i:62696;b:1;i:62701;b:1;i:62719;b:1;i:62721;b:1;i:62720;b:1;i:62722;b:1;i:62723;b:1;i:62476;b:1;i:62749;b:1;i:62733;i:1;i:62739;i:0;i:62756;b:1;i:62760;b:1;i:62740;b:1;i:62702;b:1;i:62776;i:1;i:62777;b:1;i:62794;b:1;i:62798;i:1;i:62796;i:1;i:62805;b:1;i:62808;b:1;i:62797;i:0;i:62819;b:1;i:62826;b:1;i:62824;b:1;i:62713;b:1;i:62829;b:1;i:62833;b:1;i:62838;b:1;i:62840;b:1;i:62842;b:1;i:62828;b:1;i:62845;b:1;i:62846;b:1;i:62811;b:1;i:62848;b:1;i:62850;b:1;i:62861;b:1;i:62863;b:1;i:62870;b:1;i:62875;b:1;i:62888;b:1;i:62887;b:1;i:62898;b:1;i:62885;i:1;i:62894;i:1;i:62900;b:1;i:62906;b:1;i:62891;b:1;i:62916;b:1;i:62918;b:1;i:62868;i:1;i:62867;i:1;i:62904;i:0;i:62909;i:0;i:62872;i:1;i:62927;b:1;i:62878;b:1;i:62890;b:1;i:62905;b:1;i:62929;b:1;i:62939;b:1;i:62941;b:1;i:62944;i:1;i:62926;i:1;i:62948;b:1;i:62952;i:1;i:62928;i:0;i:62954;b:1;i:62955;b:1;i:62953;b:1;i:62959;b:1;i:62958;b:1;i:62968;b:1;i:62957;i:1;i:62956;b:1;i:62821;b:1;i:62849;b:1;i:62986;b:1;i:62964;b:1;i:62991;b:1;i:62999;b:1;i:62973;b:1;i:62993;b:1;i:63003;b:1;i:62994;b:1;i:63017;b:1;i:63016;b:1;i:63019;b:1;i:63018;b:1;i:63021;b:1;i:63025;b:1;i:63028;b:1;i:63027;b:1;i:63030;b:1;i:63034;i:1;i:63035;i:0;i:63024;i:1;i:63026;i:0;i:63032;b:1;i:63044;i:1;i:63055;i:0;i:63057;i:0;i:63042;i:0;i:63036;b:1;i:63059;b:1;i:63066;i:1;i:63058;i:1;i:63069;i:1;i:63073;b:1;i:63077;b:1;i:63072;b:1;i:63078;b:1;i:63083;b:1;i:63082;b:1;i:63085;b:1;i:63084;b:1;i:63086;b:1;i:63087;b:1;i:63090;i:1;i:63091;b:1;i:63079;b:1;i:63102;b:1;i:63101;i:1;i:63103;i:0;i:63104;i:0;i:63089;b:1;i:63111;i:1;i:63113;i:1;i:63116;i:1;i:63117;b:1;i:63126;b:1;i:63131;b:1;i:63125;b:1;i:62984;b:1;i:63136;b:1;i:63122;b:1;i:63121;b:1;i:63139;b:1;i:63093;b:1;i:63061;b:1;i:63148;b:1;i:63149;b:1;i:63124;i:1;i:63146;i:0;i:63154;i:0;i:63152;i:0;i:63165;b:1;i:63164;b:1;i:63166;b:1;i:63168;b:1;i:63147;b:1;i:62923;i:1;i:63157;i:1;i:63175;b:1;i:63177;b:1;i:63163;b:1;i:63196;b:1;i:63198;b:1;i:63200;b:1;i:63155;b:1;i:63160;b:1;i:63204;b:1;i:63191;i:1;i:63194;i:0;i:63199;i:0;i:63201;i:0;i:63180;i:1;i:63181;i:0;i:63182;i:0;i:63212;b:1;i:63210;b:1;i:63219;i:1;i:63193;b:1;i:63217;b:1;i:63213;b:1;i:63241;b:1;i:63246;b:1;i:63240;b:1;i:63248;b:1;i:63242;b:1;i:63259;b:1;i:63258;b:1;i:63265;b:1;i:63247;b:1;i:63267;i:1;i:63244;b:1;i:63269;b:1;i:63225;b:1;i:63279;b:1;i:63281;b:1;i:63287;b:1;i:63289;b:1;i:63298;b:1;i:63299;b:1;i:63301;b:1;i:63300;b:1;i:63255;b:1;i:63297;b:1;i:63309;b:1;i:63308;b:1;i:63317;b:1;i:63323;b:1;i:63294;i:0;i:63330;b:1;i:63331;b:1;i:63334;i:1;i:63333;i:1;i:63337;b:1;i:63336;b:1;i:63341;b:1;i:63348;b:1;i:63351;b:1;i:63352;b:1;i:63358;b:1;i:63335;i:1;i:63340;i:0;i:63345;i:0;i:63346;i:0;i:63364;i:0;i:63362;i:0;i:63368;b:1;i:63369;b:1;i:63356;b:1;i:63373;b:1;i:63158;b:1;i:63313;b:1;i:63383;b:1;i:63393;b:1;i:63379;i:1;i:63380;i:0;i:63381;i:0;i:63382;i:0;i:63385;i:0;i:63386;i:0;i:63396;i:0;i:63397;i:0;i:54350;b:1;i:63378;b:1;i:63390;b:1;i:63409;b:1;i:63354;b:1;i:63417;b:1;i:63419;b:1;i:63387;b:1;i:63423;b:1;i:63403;b:1;i:63428;b:1;i:63435;b:1;i:63436;b:1;i:63434;b:1;i:63405;i:1;i:63407;i:1;i:63443;b:1;i:63446;b:1;i:63455;b:1;i:63457;b:1;i:63430;i:1;i:63449;i:1;i:63415;i:0;i:63460;b:1;i:63468;b:1;i:63470;b:1;i:63471;b:1;i:63464;b:1;i:63475;b:1;i:63473;b:1;i:63474;i:1;i:63476;i:0;i:60267;b:1;i:63486;b:1;i:63485;b:1;i:63488;b:1;i:63492;b:1;i:63411;b:1;i:63499;b:1;i:63494;b:1;i:63463;b:1;i:63506;b:1;i:63504;b:1;i:63509;b:1;i:63516;i:1;i:63511;b:1;i:63530;b:1;i:63515;b:1;i:63533;b:1;i:63532;b:1;i:63514;b:1;i:63519;b:1;i:63483;b:1;i:63529;i:1;i:63538;b:1;i:63537;b:1;i:63545;b:1;i:63541;i:0;i:63552;b:1;i:63561;b:1;i:63562;b:1;i:63557;i:0;i:63556;i:0;i:63551;b:1;i:63564;b:1;i:63582;b:1;i:63567;i:1;i:63568;i:0;i:63569;i:0;i:63574;i:0;i:63576;i:0;i:63575;i:0;i:63559;b:1;i:63592;i:1;i:63570;b:1;i:63606;b:1;i:63612;i:1;i:63613;i:1;i:63614;i:1;i:63598;i:1;i:63604;i:0;i:63605;i:0;i:63607;i:0;i:63608;i:0;i:63616;i:0;i:63617;i:0;i:63619;i:0;i:63620;i:0;i:63602;i:0;i:62924;b:1;i:63632;i:1;i:63634;i:1;i:63635;i:1;i:63691;b:1;i:63690;b:1;i:63696;b:1;i:63644;i:1;i:63680;i:1;i:63698;i:0;i:63618;i:1;i:63711;b:1;i:63725;b:1;i:63664;b:1;i:63734;i:1;i:63741;b:1;i:63752;b:1;i:63758;b:1;i:1471;b:0;i:63630;b:1;i:63755;b:1;i:63745;b:1;i:63754;b:1;i:63718;b:1;i:63770;b:1;i:63774;b:1;i:63784;b:1;i:63721;i:1;i:63732;i:0;i:63723;i:0;i:63724;i:0;i:63733;i:0;i:63735;i:0;i:63736;i:0;i:63737;i:0;i:63738;i:0;i:63739;i:0;i:63740;i:0;i:63761;i:0;i:63757;i:1;i:63756;i:0;i:63753;i:0;i:63773;i:0;i:63763;i:0;i:63776;i:0;i:63783;i:0;i:63785;i:0;i:63764;i:0;i:63742;i:0;i:63450;b:1;i:63799;b:1;i:63802;b:1;i:63800;b:1;i:63801;b:1;i:63804;b:1;i:63808;b:1;i:63806;b:1;i:63787;b:1;i:17562;b:1;i:63821;b:1;i:63823;b:1;i:63822;b:1;i:63828;b:1;i:63824;b:1;i:63835;b:1;i:63842;b:1;i:63849;b:1;i:63844;b:1;i:63878;b:1;i:63779;b:1;i:63833;b:1;i:63920;b:1;i:63921;b:1;i:63923;b:1;i:63930;b:1;i:63932;b:1;i:63944;b:1;i:63949;b:1;i:63950;b:1;i:63952;b:1;i:63955;b:1;i:63958;b:1;i:64007;b:1;i:64029;b:1;i:64031;b:1;i:64033;b:1;i:64040;b:1;i:64045;b:1;i:64043;b:1;i:64037;b:1;i:63913;b:1;i:64052;b:1;i:64065;b:1;i:64064;b:1;i:64067;b:1;i:64062;b:1;i:64066;b:1;i:63941;b:1;i:64048;b:1;i:64051;b:1;i:64060;b:1;i:64069;i:1;i:64072;i:1;i:63853;i:1;i:63886;i:0;i:64046;i:0;i:64070;i:0;i:64041;i:0;i:64058;b:1;i:64083;b:1;i:64084;b:1;i:64018;b:1;i:64088;b:1;i:64089;b:1;i:64042;b:1;i:64099;b:1;i:64091;b:1;i:64102;b:1;i:64093;i:0;i:64076;b:1;i:64080;b:1;i:64113;b:1;i:64108;i:1;i:64095;i:0;i:64100;i:0;i:64107;i:0;i:64110;i:1;i:64122;i:0;i:64123;i:0;i:64124;i:0;i:64125;i:0;i:64126;i:0;i:63820;i:0;i:63827;i:1;i:63840;i:0;i:63847;i:0;i:63855;i:0;i:63856;i:0;i:63857;i:0;i:63858;i:0;i:63860;i:0;i:63861;i:0;i:64119;b:1;i:64118;b:1;i:63862;i:0;i:63863;i:0;i:63864;i:0;i:64086;b:1;i:63865;i:0;i:63866;i:1;i:63867;i:0;i:63868;i:0;i:63869;i:1;i:64114;b:1;i:64145;b:1;i:63870;i:0;i:63871;i:0;i:63872;i:0;i:63873;i:0;i:63874;i:0;i:63877;i:0;i:63879;i:0;i:63881;i:0;i:63882;i:0;i:64151;b:1;i:64155;b:1;i:64139;b:1;i:64158;b:1;i:64143;b:1;i:64160;b:1;i:64154;b:1;i:64101;b:1;i:64166;b:1;i:64161;b:1;i:63883;i:0;i:63885;i:0;i:63887;i:0;i:63888;i:0;i:64112;b:1;i:63889;i:0;i:63890;i:1;i:63894;i:0;i:63895;i:1;i:63896;i:0;i:63902;i:0;i:63904;i:1;i:64146;i:1;i:64150;i:1;i:64152;i:0;i:64153;i:0;i:64096;b:1;i:64156;i:1;i:64207;b:1;i:64208;b:1;i:64209;b:1;i:64211;b:1;i:64212;b:1;i:64214;b:1;i:64198;b:1;i:64219;b:1;i:64210;b:1;i:64225;b:1;i:64228;b:1;i:64237;b:1;i:64238;b:1;i:64255;b:1;i:64257;b:1;i:64261;b:1;i:64265;b:1;i:64224;b:1;i:64275;b:1;i:64278;b:1;i:64280;b:1;i:64282;b:1;i:64253;b:1;i:64252;b:1;i:64239;b:1;i:64240;b:1;i:64241;b:1;i:64242;b:0;i:64243;b:0;i:64244;b:0;i:64245;b:0;i:64246;b:1;i:64251;b:1;i:64247;b:0;i:64248;b:1;i:64249;b:0;i:64250;b:0;i:64235;b:1;i:64283;b:1;i:64296;i:0;i:64297;b:1;i:64284;b:1;i:64310;b:1;i:64313;b:1;i:64299;b:1;i:64314;b:1;i:64311;b:1;i:64318;b:1;i:64325;b:1;i:64326;b:1;i:64321;b:1;i:64329;b:1;i:64305;b:1;i:64332;b:1;i:64339;i:1;i:64334;b:1;i:64328;b:1;i:64342;b:1;i:64347;b:1;i:64346;b:1;i:64320;b:1;i:64348;b:1;i:64349;b:1;i:64292;b:1;i:64547;b:1;i:64550;b:1;i:64553;b:1;i:64350;b:1;i:64554;b:1;i:64526;b:1;i:64546;b:1;i:64544;b:1;i:64345;b:1;i:64331;b:1;i:64490;b:1;i:64341;b:1;i:64595;b:1;i:64582;b:1;i:64616;b:1;i:64618;b:1;i:64620;b:1;i:64449;b:1;i:64460;b:1;i:64683;b:1;i:64685;b:1;i:64688;b:1;i:64705;b:1;i:64712;b:1;i:64708;b:1;i:64724;b:1;i:64723;b:1;i:64731;b:1;i:64736;b:1;i:64746;b:1;i:64756;b:1;i:64651;b:1;i:64690;b:1;i:64752;b:1;i:64281;b:1;i:64750;b:1;i:64775;b:1;i:64763;b:1;i:64781;b:1;i:64653;b:1;i:64790;b:1;i:64797;b:1;i:64811;b:1;i:64812;b:1;i:64814;b:1;i:64773;b:1;i:64351;b:1;i:64733;b:1;i:64729;b:1;i:64782;b:1;i:64824;b:1;i:64828;b:1;i:64827;b:1;i:64830;b:1;i:64016;b:1;i:64822;b:1;i:64855;b:1;i:64862;b:1;i:64846;b:1;i:64831;b:1;i:64829;b:1;i:64869;b:1;i:64867;b:1;i:64871;b:1;i:64874;b:1;i:64882;b:1;i:64896;b:1;i:64803;b:1;i:64899;b:1;i:64898;b:1;i:64900;b:1;i:64918;b:1;i:64922;b:1;i:64921;b:1;i:64926;b:1;i:64937;b:1;i:64940;b:1;i:64944;b:1;i:64946;b:1;i:64943;b:1;i:64964;b:1;i:64966;b:1;i:64965;b:1;i:64970;b:1;i:64973;b:1;i:64977;b:1;i:64980;b:1;i:64971;b:1;i:64994;b:1;i:64996;b:1;i:64969;b:1;i:64989;b:1;i:65000;b:1;i:65013;b:1;i:65014;b:1;i:65010;b:1;i:64231;b:1;i:64993;b:1;i:65018;b:1;i:65021;b:1;i:64955;b:1;i:65024;b:1;i:64979;b:1;i:65058;b:1;i:65056;b:1;i:65062;b:1;i:65063;b:1;i:65042;b:1;i:65040;b:1;i:65075;b:1;i:65078;b:1;i:65076;b:1;i:65087;b:1;i:65099;b:1;i:65098;b:1;i:65026;b:1;i:65104;b:1;i:65095;b:1;i:65107;b:1;i:65090;b:1;i:65085;b:1;i:65061;b:1;i:65113;b:1;i:65106;b:1;i:65110;b:1;i:65109;b:1;i:65108;b:1;i:65120;b:1;i:65119;b:1;i:65126;b:1;i:65032;b:1;i:65007;b:1;i:65145;b:1;i:65144;b:1;i:65149;b:1;i:65155;b:1;i:65157;b:1;i:65138;b:1;i:65160;b:1;i:65151;b:1;i:65164;b:1;i:65153;b:1;i:65163;b:1;i:65179;b:1;i:65178;b:1;i:65052;b:1;i:65180;b:1;i:65196;b:1;i:65188;b:1;i:65199;b:1;i:65212;b:1;i:65217;b:1;i:65218;b:1;i:65205;b:1;i:65224;b:1;i:65227;b:1;i:65214;b:1;i:65234;b:1;i:65241;b:1;i:65162;b:1;i:65243;b:1;i:65247;b:1;i:65246;b:1;i:65253;b:1;i:65251;b:1;i:65254;b:1;i:65271;b:1;i:65273;b:1;i:65268;b:1;i:65280;b:1;i:65261;b:1;i:65281;b:1;i:65288;b:1;i:65279;b:1;i:65262;b:1;i:65305;b:1;i:65287;b:1;i:65314;b:1;i:65320;b:1;i:65310;b:1;i:65289;b:1;i:65307;b:1;i:65329;b:1;i:65335;b:1;i:65321;b:1;i:65336;b:1;i:65296;b:1;i:65294;b:1;i:65318;b:1;i:65337;b:1;i:65327;b:1;i:65360;b:1;i:65367;b:1;i:65376;b:1;i:65371;b:1;i:65381;b:1;i:65359;b:1;i:65387;b:1;i:65362;b:1;i:65400;b:1;i:65401;b:1;i:65299;b:1;i:65357;b:1;i:65412;b:1;i:65409;b:1;i:65358;b:1;i:65408;b:1;i:65417;b:1;i:65425;b:1;i:65426;b:1;i:65345;b:1;i:65427;b:1;i:65429;b:1;i:65222;b:1;i:65419;b:1;i:65432;b:1;i:65436;b:1;i:65443;b:1;i:65444;b:1;i:65445;b:1;i:65453;b:1;i:65454;b:1;i:65448;b:1;i:65458;b:1;i:65462;b:1;i:65470;b:1;i:65461;b:1;i:65402;b:1;i:65475;b:1;i:65476;b:1;i:65482;b:1;i:65487;b:1;i:65491;b:1;i:65488;b:1;i:65463;b:1;i:65422;b:1;i:65510;b:1;i:65511;b:1;i:65513;b:1;i:65515;b:1;i:65516;b:1;i:65518;b:1;i:65512;b:1;i:65526;b:1;i:65528;b:1;i:65534;b:1;i:65541;b:1;i:65540;b:1;i:65551;b:1;i:65552;b:1;i:65554;b:1;i:65558;b:1;i:65562;b:1;i:65563;b:1;i:65560;b:1;i:65566;b:1;i:65523;b:1;i:65572;b:1;i:65575;b:1;i:70734;b:1;i:70724;b:1;i:70754;b:1;i:70739;b:1;i:70729;b:1;i:70653;b:1;i:70588;b:1;i:70580;b:1;i:70750;b:1;i:70765;b:1;i:67654;b:1;i:70378;b:1;i:70613;b:1;i:67411;b:1;i:70688;b:1;i:70797;b:1;i:70798;b:1;i:70667;b:1;i:70805;b:1;i:70817;b:1;i:70823;b:1;i:70743;b:1;i:70834;b:1;i:69124;b:1;i:70848;b:1;i:70428;b:1;i:70488;b:1;i:70826;b:1;i:70722;b:1;i:70616;b:1;i:70677;b:1;i:70491;b:1;i:70598;b:1;i:70860;b:1;i:70831;b:1;i:70863;b:1;i:70880;b:1;i:70843;b:1;i:70879;b:1;i:70878;b:1;i:70877;b:1;i:70890;b:1;i:70891;b:1;i:70883;b:1;i:70819;b:1;i:70904;b:1;i:70909;b:1;i:70916;b:1;i:70845;b:1;i:70913;b:1;i:70658;b:1;i:70601;b:1;i:70908;b:1;i:70924;b:1;i:70941;b:1;i:70928;b:1;i:70590;b:1;i:70943;b:1;i:70895;b:1;i:70901;b:1;i:70937;b:1;i:70949;b:1;i:70929;b:1;i:70956;b:1;i:70957;b:1;i:70853;b:1;i:70939;b:1;i:70966;b:1;i:70960;b:1;i:70959;b:1;i:70971;b:1;i:70979;b:1;i:70978;b:1;i:70967;b:1;i:70992;b:1;i:70997;b:1;i:71000;b:1;i:71001;b:1;i:71016;b:1;i:71015;b:1;i:70998;b:1;i:70986;b:1;i:71021;b:1;i:70963;b:1;i:71030;b:1;i:71041;b:1;i:71058;b:1;i:71051;b:1;i:71048;b:1;i:71061;b:1;i:71045;b:1;i:71070;b:1;i:71042;b:1;i:71080;b:1;i:71074;b:1;i:71094;b:1;i:71052;b:1;i:71098;b:1;i:71099;b:1;i:71082;b:1;i:71107;b:1;i:71113;b:1;i:71111;b:1;i:71128;b:1;i:71124;b:1;i:71132;b:1;i:71120;b:1;i:71140;b:1;i:71125;b:1;i:71143;b:1;i:71147;b:1;i:71145;b:1;i:71084;b:1;i:71153;b:1;i:71154;b:1;i:71158;b:1;i:71159;b:1;i:71161;b:1;i:71162;b:1;i:71176;b:1;i:71169;b:1;i:71181;b:1;i:71115;b:1;i:71194;b:1;i:71187;b:1;i:71185;b:1;i:71067;b:1;i:71218;b:1;i:71217;b:1;i:71220;b:1;i:71219;b:1;i:71221;b:1;i:71227;b:1;i:71156;b:1;i:71239;b:1;i:71238;b:1;i:71229;b:1;i:71261;b:1;i:71260;b:1;i:71240;b:1;i:71277;b:1;i:71275;b:1;i:71288;b:1;i:71291;b:1;i:71290;b:1;i:71302;b:1;i:71300;b:1;i:71308;b:1;i:71279;b:1;i:71311;b:1;i:71303;b:1;i:71315;b:1;i:71305;b:1;i:71319;b:1;i:71304;b:1;i:71320;b:1;i:71316;b:1;i:71312;b:1;i:71310;b:1;i:71331;b:1;i:71336;b:1;i:71337;b:1;i:71341;b:1;i:71330;b:1;i:71282;b:1;i:71199;b:1;i:71342;b:1;i:71352;b:1;i:71223;b:1;i:71356;b:1;i:71360;b:1;i:71363;b:1;i:71343;b:1;i:71297;b:1;i:71366;b:1;i:71364;b:1;i:71358;b:1;i:71368;b:1;i:71375;b:1;i:71382;b:1;i:71387;b:1;i:71386;b:1;i:71392;b:1;i:71406;b:1;i:71388;b:1;i:71394;b:1;i:71393;b:1;i:71408;b:1;i:71420;b:1;i:71427;b:1;i:71441;b:1;i:71455;b:1;i:71462;b:1;i:71449;b:1;i:71481;b:1;i:71373;b:1;i:71487;b:1;i:71478;b:1;i:71494;b:1;i:71385;b:1;i:71495;b:1;i:71507;b:1;i:71510;b:1;i:71501;b:1;i:71514;b:1;i:71517;b:1;i:71527;b:1;i:71530;b:1;i:71532;b:1;i:71529;b:1;i:71536;b:1;i:71540;b:1;i:71546;b:1;i:71558;b:1;i:71569;b:1;i:71447;b:1;i:48459;b:1;i:71578;b:1;i:71584;b:1;i:71588;b:1;i:71601;b:1;i:71598;b:1;i:71609;b:1;i:71611;b:1;i:71543;b:1;i:71619;b:1;i:71624;b:1;i:71634;b:1;i:71605;b:1;i:71630;b:1;i:71654;b:1;i:71656;b:1;i:71655;b:1;i:71660;b:1;i:71671;b:1;i:71653;b:1;i:71676;b:1;i:71678;b:1;i:71681;b:1;i:71702;b:1;i:71704;b:1;i:71707;b:1;i:71692;b:1;i:71728;b:1;i:71737;b:1;i:71745;b:1;i:71726;b:1;i:71742;b:1;i:71743;b:1;i:71759;b:1;i:71768;b:1;i:71741;b:1;i:71751;b:1;i:71783;b:1;i:71746;b:1;i:71754;b:1;i:71812;b:1;i:71815;b:1;i:71814;b:1;i:71818;b:1;i:71820;b:1;i:71786;b:1;i:71756;b:1;i:71827;b:1;i:71835;b:1;i:71836;b:1;i:71838;b:1;i:71842;b:1;i:71844;b:1;i:71850;b:1;i:71855;b:1;i:71854;b:1;i:71892;b:1;i:71852;b:1;i:71898;b:1;i:71901;b:1;i:71900;b:1;i:71909;b:1;i:71912;b:1;i:71914;b:1;i:71907;b:1;i:71919;b:1;i:71910;b:1;i:71887;b:1;i:71932;b:1;i:71931;b:1;i:71872;b:1;i:70495;i:1;i:71947;b:1;i:71951;b:1;i:71955;b:1;i:71938;b:1;i:71963;b:1;i:71971;b:1;i:71970;b:1;i:71968;b:1;i:71987;b:1;i:71960;b:1;i:71991;b:1;i:71993;b:1;i:72000;b:1;i:72002;b:1;i:72008;b:1;i:72014;b:1;i:71992;b:1;i:71979;b:1;i:71934;b:1;i:71744;b:1;i:71895;b:1;i:72035;b:1;i:72034;b:1;i:72033;b:1;i:72032;b:1;i:71962;b:1;i:72061;b:1;i:72062;b:1;i:72072;b:1;i:72071;b:1;i:71864;b:1;i:71990;b:1;i:72078;b:1;i:72082;b:1;i:72090;b:1;i:72094;b:1;i:72104;b:1;i:72107;b:1;i:72108;b:1;i:72109;b:1;i:72112;b:1;i:72123;b:1;i:72126;b:1;i:72127;b:1;i:72121;b:1;i:72133;b:1;i:72106;b:1;i:72154;b:1;i:72155;b:1;i:72161;b:1;i:72134;b:1;i:72122;b:1;i:72169;b:1;i:72160;b:1;i:72170;b:1;i:72179;b:1;i:72163;b:1;i:72115;b:1;i:72201;b:1;i:72053;b:1;i:72192;b:1;i:72124;b:1;i:72218;b:1;i:72219;b:1;i:72229;b:1;i:72234;b:1;i:72237;b:1;i:72190;b:1;i:72200;b:1;i:72185;b:1;i:72242;b:1;i:72233;b:1;i:72244;b:1;i:72257;b:1;i:72215;b:1;i:72236;b:1;i:72260;b:1;i:72259;b:1;i:72271;b:1;i:72216;b:1;i:72278;b:1;i:72036;b:1;i:72276;b:1;i:72180;b:1;i:72188;b:1;i:72288;b:1;i:72301;b:1;i:72296;b:1;i:72305;b:1;i:72326;b:1;i:72328;b:1;i:72327;b:1;i:72329;b:1;i:72331;b:1;i:72339;b:1;i:72324;b:1;i:72269;b:1;i:72087;b:1;i:72083;b:1;i:72319;b:1;i:72358;b:1;i:72361;b:1;i:72356;b:1;i:72395;b:1;i:72398;b:1;i:72399;b:1;i:72404;b:1;i:72102;b:1;i:72318;b:1;i:72413;b:1;i:72340;b:1;i:72338;b:1;i:72414;b:1;i:72425;b:1;i:72393;b:1;i:72448;b:1;i:72465;b:1;i:72280;b:1;i:72464;b:1;i:72474;b:1;i:72478;b:1;i:72495;b:1;i:72497;b:1;i:72455;b:1;i:72440;b:1;i:72458;b:1;i:72508;b:1;i:72514;b:1;i:72515;b:1;i:72510;b:1;i:72471;b:1;i:72506;b:1;i:72535;b:1;i:72542;b:1;i:72545;b:1;i:72549;b:1;i:72550;b:1;i:72442;b:1;i:72496;b:1;i:72360;b:1;i:72494;b:1;i:72555;b:1;i:72499;b:1;i:72519;b:1;i:72566;b:1;i:72585;b:1;i:72586;b:1;i:72602;b:1;i:72548;b:1;i:72563;b:1;i:72622;b:1;i:72532;b:1;i:72536;b:1;i:72569;b:1;i:72623;b:1;i:72578;b:1;i:72587;b:1;i:72591;b:1;i:72598;b:1;i:72245;b:1;i:72597;b:1;i:72616;b:1;i:72609;b:1;i:72572;b:1;i:72540;b:1;i:72435;b:1;i:72652;b:1;i:72669;b:1;i:72417;b:1;i:72682;b:1;i:72625;b:1;i:72661;b:1;i:72683;b:1;i:72688;b:1;i:72644;b:1;i:72703;b:1;i:72678;b:1;i:72702;b:1;i:72718;b:1;i:72687;b:1;i:72723;b:1;i:72698;b:1;i:72696;b:1;i:72730;b:1;i:72745;b:1;i:72680;b:1;i:72711;b:1;i:72758;b:1;i:72759;b:1;i:72774;b:1;i:72754;b:1;i:72760;b:1;i:72776;b:1;i:72780;b:1;i:72783;b:1;i:72781;b:1;i:72620;b:1;i:72560;b:1;i:72794;b:1;i:72793;b:1;i:72792;b:1;i:72795;b:1;i:72767;b:1;i:72817;b:1;i:72808;b:1;i:72819;b:1;i:72826;b:1;i:72829;b:1;i:72834;b:1;i:72837;b:1;i:72840;b:1;i:72850;b:1;i:72860;b:1;i:72849;b:1;i:72859;b:1;i:72861;b:1;i:72835;b:1;i:72871;b:1;i:72831;b:1;i:72846;b:1;i:72886;b:1;i:72901;b:1;i:72933;b:1;i:72942;b:1;i:72948;b:1;i:72936;b:1;i:72953;b:1;i:72922;b:1;i:72960;b:1;i:72749;b:1;i:72895;b:1;i:72770;b:1;i:72778;b:1;i:72825;b:1;i:72951;b:1;i:72900;b:1;i:72914;b:1;i:72912;b:1;i:72959;b:1;i:72932;b:1;i:72916;b:1;i:72915;b:1;i:72945;b:1;i:72946;b:1;i:72965;b:1;i:72976;b:1;i:72981;b:1;i:72983;b:1;i:72947;b:1;i:72949;b:1;i:72994;b:1;i:73011;b:1;i:72882;b:1;i:73019;b:1;i:73018;b:1;i:72930;b:1;i:73020;b:1;i:72935;b:1;i:73010;b:1;i:72929;b:1;i:72928;b:1;i:72854;b:1;i:73033;b:1;i:73032;b:1;i:73016;b:1;i:73037;b:1;i:73061;b:1;i:73060;b:1;i:72990;b:1;i:73034;b:1;i:73028;b:1;i:73041;b:1;i:73047;b:1;i:73056;b:1;i:73050;b:1;i:73079;b:1;i:73091;b:1;i:73052;b:1;i:73086;b:1;i:73099;b:1;i:73119;b:1;i:73121;b:1;i:73142;b:1;i:73149;b:1;i:73082;b:1;i:73112;b:1;i:73157;b:1;i:73167;b:1;i:73177;b:1;i:73179;b:1;i:73183;b:1;i:73066;b:1;i:73088;b:1;i:73137;b:1;i:73147;b:1;i:73150;b:1;i:73163;b:1;i:73170;b:1;i:73201;b:1;i:73144;b:1;i:73153;b:1;i:73208;b:1;i:73211;b:1;i:73216;b:1;i:73217;b:1;i:73229;b:1;i:73233;b:1;i:73235;b:1;i:73223;b:1;i:73240;b:1;i:73245;b:1;i:73259;b:1;i:73256;b:1;i:73244;b:1;i:73054;b:1;i:73198;b:1;i:73249;b:1;i:73266;b:1;i:73274;b:1;i:72926;b:1;i:73193;b:1;i:73196;b:1;i:73251;b:1;i:73102;b:1;i:73280;b:1;i:73279;b:1;i:73268;b:1;i:73285;b:1;i:73261;b:1;i:73264;b:1;i:73292;b:1;i:73295;b:1;i:73289;b:1;i:73300;b:1;i:73308;b:1;i:73309;b:1;i:73323;b:1;i:73322;b:1;i:73331;b:1;i:73369;b:1;i:73373;b:1;i:73374;b:1;i:73368;b:1;i:73381;b:1;i:73371;b:1;i:73370;b:1;i:73357;b:1;i:73389;b:1;i:73367;b:1;i:73394;b:1;i:73344;b:1;i:73359;b:1;i:73398;b:1;i:73404;b:1;i:73396;b:1;i:73306;b:1;i:73281;b:1;i:73419;b:1;i:73038;b:1;i:73200;b:1;i:73284;b:1;i:73297;b:1;i:73349;b:1;i:73334;b:1;i:73433;b:1;i:73443;b:1;i:73448;b:1;i:73457;b:1;i:73451;b:1;i:73441;b:1;i:73302;b:1;i:73428;b:1;i:73431;b:1;i:73460;b:1;i:73463;b:1;i:73459;b:1;i:73486;b:1;i:73487;b:1;i:73475;b:1;i:73437;b:1;i:73506;b:1;i:73514;b:1;i:73440;b:1;i:73517;b:1;i:73545;b:1;i:73542;b:1;i:73477;b:1;i:73515;b:1;i:73488;b:1;i:73494;b:1;i:73526;b:1;i:73543;b:1;i:73567;b:1;i:73525;b:1;i:73575;b:1;i:73583;b:1;i:73584;b:1;i:73531;b:1;i:73585;b:1;i:73579;b:1;i:73540;b:1;i:73571;b:1;i:73602;b:1;i:73590;b:1;i:73599;b:1;i:73610;b:1;i:73611;b:1;i:73580;b:1;i:73573;b:1;i:73519;b:1;i:73578;b:1;i:73615;b:1;i:73621;b:1;i:73614;b:1;i:73629;b:1;i:73627;b:1;i:73631;b:1;i:73619;b:1;i:73652;b:1;i:73655;b:1;i:73662;b:1;i:73630;b:1;i:73638;b:1;i:73661;b:1;i:73663;b:1;i:73666;b:1;i:73668;b:1;i:73670;b:1;i:73685;b:1;i:73679;b:1;i:73683;b:1;i:73688;b:1;i:73659;b:1;i:73651;b:1;i:73698;b:1;i:73724;b:1;i:73727;b:1;i:73733;b:1;i:73722;b:1;i:73708;b:1;i:73717;b:1;i:73739;b:1;i:73743;b:1;i:73744;b:1;i:73735;b:1;i:73715;b:1;i:73737;b:1;i:73754;b:1;i:73695;b:1;i:73758;b:1;i:73757;b:1;i:73763;b:1;i:73769;b:1;i:73772;b:1;i:73694;b:1;i:73765;b:1;i:73764;b:1;i:73796;b:1;i:73798;b:1;i:73802;b:1;i:73797;b:1;i:73803;b:1;i:73752;b:1;i:73786;b:1;i:73789;b:1;i:73788;b:1;i:73809;b:1;i:73810;b:1;i:73808;b:1;i:73839;b:1;i:73836;b:1;i:73842;b:1;i:73795;b:1;i:73760;b:1;i:73781;b:1;i:73816;b:1;i:73834;b:1;i:73868;b:1;i:73866;b:1;i:73867;b:1;i:73878;b:1;i:73880;b:1;i:73881;b:1;i:73673;b:1;i:73658;b:1;i:73654;b:1;i:73826;b:1;i:73822;b:1;i:73858;b:1;i:73849;b:1;i:73902;b:1;i:73899;b:1;i:73832;b:1;i:73918;b:1;i:73908;b:1;i:73933;b:1;i:73945;b:1;i:73946;b:1;i:73906;b:1;i:73928;b:1;i:73915;b:1;i:73922;b:1;i:73934;b:1;i:73951;b:1;i:73937;b:1;i:73716;b:1;i:73984;b:1;i:73981;b:1;i:73949;b:1;i:74008;b:1;i:73989;b:1;i:73896;b:1;i:73885;b:1;i:73884;b:1;i:73964;b:1;i:74021;b:1;i:74016;b:1;i:74041;b:1;i:74024;b:1;i:73995;b:1;i:74045;b:1;i:74051;b:1;i:74050;b:1;i:74056;b:1;i:74066;b:1;i:74067;b:1;i:74080;b:1;i:74078;b:1;i:74086;b:1;i:74058;b:1;i:74094;b:1;i:74088;b:1;i:74099;b:1;i:74101;b:1;i:74065;b:1;i:74061;b:1;i:74097;b:1;i:74026;b:1;i:74126;b:1;i:74132;b:1;i:74138;b:1;i:74142;b:1;i:74161;b:1;i:74165;b:1;i:74171;b:1;i:74180;b:1;i:74172;b:1;i:74069;b:1;i:74198;b:1;i:74113;b:1;i:74117;b:1;i:74166;b:1;i:74175;b:1;i:74215;b:1;i:74221;b:1;i:74195;b:1;i:74231;b:1;i:74236;b:1;i:74238;b:1;i:74219;b:1;i:74228;b:1;i:74265;b:1;i:74264;b:1;i:74274;b:1;i:74271;b:1;i:74162;b:1;i:74199;b:1;i:74229;b:1;i:74254;b:1;i:74190;b:1;i:74279;b:1;i:74314;b:1;i:74284;b:1;i:74283;b:1;i:74316;b:1;i:74320;b:1;i:74326;b:1;i:74335;b:1;i:74356;b:1;i:74373;b:1;i:74379;b:1;i:74392;b:1;i:74402;b:1;i:74404;b:1;i:74438;b:1;i:74443;b:1;i:74450;b:1;i:74452;b:1;i:74394;b:1;i:74281;b:1;i:74306;b:1;i:74340;b:1;i:74464;b:1;i:74465;b:1;i:74453;b:1;i:74467;b:1;i:74505;b:1;i:74514;b:1;i:74524;b:1;i:74528;b:1;i:74571;b:1;i:74582;b:1;i:74595;b:1;i:74608;b:1;i:74594;b:1;i:74609;b:1;i:74639;b:1;i:74637;b:1;i:74642;b:1;i:74647;b:1;i:74651;b:1;i:74649;b:1;i:74662;b:1;i:74661;b:1;i:74650;b:1;i:74645;b:1;i:74640;b:1;i:74665;b:1;i:74668;b:1;i:74666;b:1;i:74667;b:1;i:74615;b:1;i:74610;b:1;i:74675;b:1;i:74671;b:1;i:74446;b:1;i:74701;b:1;i:74684;b:1;i:74718;b:1;i:74625;b:1;i:74722;b:1;i:74727;b:1;i:74717;b:1;i:73588;b:1;i:74003;b:1;i:74001;b:1;i:74083;b:1;i:74124;b:1;i:74367;b:1;i:74462;b:1;i:74375;b:1;i:74377;b:1;i:74381;b:1;i:74384;b:1;i:74388;b:1;i:74396;b:1;i:74398;b:1;i:74439;b:1;i:74444;b:1;i:74448;b:1;i:74463;b:1;i:74471;b:1;i:74475;b:1;i:74474;b:1;i:74476;b:1;i:74478;b:1;i:74485;b:1;i:74502;b:1;i:74500;b:1;i:74503;b:1;i:74565;b:1;i:74569;b:1;i:74620;b:1;i:74624;b:1;i:74669;b:1;i:74677;b:1;i:74700;b:1;i:74702;b:1;i:74713;b:1;i:74737;b:1;i:74750;b:1;i:74755;b:1;i:74756;b:1;i:74757;b:1;i:74743;b:1;i:74760;b:1;i:74763;b:1;i:74766;b:1;i:74768;b:1;i:74765;b:1;i:74752;b:1;i:74741;b:1;i:74784;b:1;i:74788;b:1;i:74731;b:1;i:74735;b:1;i:74773;b:1;i:74794;b:1;i:74797;b:1;i:74744;b:1;i:74742;b:1;i:74729;b:1;i:74799;b:1;i:74802;b:1;i:74806;b:1;i:74622;b:1;i:74591;b:1;i:74586;b:1;i:74522;b:1;i:74778;b:1;i:74826;b:1;i:74828;b:1;i:74827;b:1;i:74829;b:1;i:74839;b:1;i:74840;b:1;i:74841;b:1;i:74842;b:1;i:74844;b:1;i:74849;b:1;i:74852;b:1;i:74860;b:1;i:74854;b:1;i:74869;b:1;i:74874;b:1;i:74875;b:1;i:74830;b:1;i:74886;b:1;i:74889;b:1;i:74888;b:1;i:74843;b:1;i:74957;b:1;i:74959;b:1;i:74960;b:1;i:74962;b:1;i:74969;b:1;i:74972;b:1;i:74982;b:1;i:74983;b:1;i:74992;b:1;i:75010;b:1;i:75029;b:1;i:75009;b:1;i:75046;b:1;i:75060;b:1;i:75063;b:1;i:74589;b:1;i:74833;b:1;i:74864;b:1;i:74863;b:1;i:74898;b:1;i:74956;b:1;i:74958;b:1;i:74980;b:1;i:75018;b:1;i:75053;b:1;i:75040;b:1;i:75037;b:1;i:75083;b:1;i:75096;b:1;i:75125;b:1;i:75097;b:1;i:75085;b:1;i:75123;b:1;i:75087;b:1;i:75151;b:1;i:75170;b:1;i:75156;b:1;i:75026;b:1;i:75153;b:1;i:75193;b:1;i:75203;b:1;i:75209;b:1;i:75215;b:1;i:75216;b:1;i:75213;b:1;i:75212;b:1;i:75738;b:1;i:75746;b:1;i:75747;b:1;i:75727;b:1;i:75745;b:1;i:75764;b:1;i:75766;b:1;i:75850;b:1;i:75855;b:1;i:75858;b:1;i:32194;b:1;i:75914;b:1;i:75919;b:1;i:75799;b:1;i:75849;b:1;i:75206;b:1;i:75163;b:1;i:76196;b:1;i:76205;b:1;i:76206;b:1;i:76226;b:1;i:76233;b:1;i:76229;b:1;i:76235;b:1;i:76343;b:1;i:76346;b:1;i:76336;b:1;i:76369;b:1;i:76385;b:1;i:76362;b:1;i:76399;b:1;i:76407;b:1;i:76405;b:1;i:76429;b:1;i:76430;b:1;i:76423;b:1;}" string(4) "i:0;" string(4) "i:0;" string(4) "i:0;" string(4) "i:0;" string(4) "i:0;" string(4) "i:0;" string(4) "i:0;" string(4) "i:0;" string(4) "i:0;" string(4) "i:0;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(15) "s:8:"disabled";" string(15) "s:8:"disabled";" string(15) "s:8:"disabled";" string(15) "s:8:"disabled";" string(15) "s:8:"disabled";" string(15) "s:8:"disabled";" string(15) "s:8:"disabled";" string(15) "s:8:"disabled";" string(15) "s:8:"disabled";" string(15) "s:8:"disabled";" string(21) "s:13:"64.191.203.34";" string(295) "s:286:"Google Yahoo BlogPulse ia_archiver Pingdom Teoma Netcraft Mnogosearch page.store libwww.perl libcurl del.icio.us wiji WebImages User-Agent: Python-urllib Twitturly Ruby facebookexternalhit ColdFusion PycURL PEAR HTTP_Request class LWP::Simple 'artviper WordPress";" string(70) "s:62:"cron.php feed rss.xml image-cacher/* iframe/refresh reset";" string(29) "s:21:"/bot|spider|crawler/i";" string(8) "s:1:"3";" string(4) "i:0;" string(8) "s:1:"3";" string(9) "s:2:"90";" string(4) "i:1;" string(4) "i:1;" string(8) "s:1:"5";" string(309) "s:300:"Hi [profile-name], We are conducting a survey, and your response would be appreciated. Here is a link to the survey: [feedback-url] This link is uniquely tied to this survey and your email address. Please do not forward this message. Thanks for your participation! The Strand NGS Team";" string(44) "s:36:"Strand NGS Customer Support Feedback";" string(8) "s:1:"3";" string(81) "s:73:"New ticket has been created [field_ticket_id-formatted]. Strand Portals";" string(9) "s:2:"Hi";" string(55) "s:47:"Escalating Ticket [ticket-id] Strand Portals";" string(9) "s:2:"Hi";" string(4) "b:1;" string(4) "b:0;" string(40) "s:32:"9fdc678f1d51968c8e1b7e83bfdbd00f";" string(4) "i:0;" string(4) "i:1;" string(4) "i:1;" string(4) "b:1;" string(10) "s:3:"600";" string(295) "a:8:{s:17:"zen_block_editing";s:1:"1";s:14:"zen_breadcrumb";s:3:"yes";s:24:"zen_breadcrumb_separator";s:5:" › ";s:19:"zen_breadcrumb_home";s:1:"1";s:23:"zen_breadcrumb_trailing";s:1:"1";s:20:"zen_breadcrumb_title";s:1:"0";s:20:"zen_rebuild_registry";s:1:"1";s:14:"zen_wireframes";s:1:"0";}" string(774) "a:25:{s:17:"zen_block_editing";i:1;s:14:"zen_breadcrumb";s:3:"yes";s:24:"zen_breadcrumb_separator";s:5:" › ";s:19:"zen_breadcrumb_home";i:1;s:23:"zen_breadcrumb_trailing";i:1;s:20:"zen_breadcrumb_title";i:0;s:20:"zen_rebuild_registry";i:1;s:14:"zen_wireframes";i:0;s:11:"toggle_logo";i:1;s:11:"toggle_name";i:0;s:13:"toggle_slogan";i:0;s:14:"toggle_mission";i:1;s:24:"toggle_node_user_picture";i:0;s:27:"toggle_comment_user_picture";i:0;s:13:"toggle_search";i:0;s:14:"toggle_favicon";i:1;s:20:"toggle_primary_links";i:0;s:22:"toggle_secondary_links";i:0;s:12:"default_logo";i:0;s:9:"logo_path";s:33:"sites/default/files/avadisimg.jpg";s:11:"logo_upload";s:0:"";s:15:"default_favicon";i:1;s:12:"favicon_path";s:0:"";s:14:"favicon_upload";s:0:"";s:5:"theme";s:8:"newtheme";}" string(2006) "a:56:{s:26:"rootcandy_navigation_icons";i:0;s:31:"rootcandy_navigation_icons_size";s:2:"24";s:24:"rootcandy_header_display";i:0;s:33:"rootcandy_navigation_custom_icons";s:0:"";s:27:"rootcandy_dashboard_display";i:0;s:24:"rootcandy_dashboard_help";s:4:"left";s:28:"rootcandy_dashboard_messages";s:5:"right";s:35:"rootcandy_dashboard_content_display";i:0;s:22:"rootcandy_help_display";i:0;s:21:"rootcandy_hide_author";i:0;s:20:"rootcandy_hide_panel";i:0;s:33:"rootcandy_navigation_source_admin";s:29:"_rootcandy_default_navigation";s:7:"mission";s:0:"";s:12:"default_logo";i:0;s:9:"logo_path";s:21:"files/crm/logo.png ";s:15:"default_favicon";i:1;s:12:"favicon_path";s:0:"";s:13:"primary_links";i:1;s:15:"secondary_links";i:1;s:11:"toggle_logo";i:0;s:14:"toggle_favicon";i:1;s:11:"toggle_name";i:1;s:13:"toggle_search";i:0;s:13:"toggle_slogan";i:0;s:14:"toggle_mission";i:1;s:24:"toggle_node_user_picture";i:0;s:27:"toggle_comment_user_picture";i:0;s:20:"toggle_primary_links";i:0;s:22:"toggle_secondary_links";i:0;s:11:"logo_upload";s:0:"";s:14:"favicon_upload";s:0:"";s:29:"rootcandy_navigation_source_1";s:0:"";s:29:"rootcandy_navigation_source_2";s:0:"";s:30:"rootcandy_navigation_source_14";s:0:"";s:30:"rootcandy_navigation_source_16";s:0:"";s:30:"rootcandy_navigation_source_13";s:0:"";s:30:"rootcandy_navigation_source_18";s:0:"";s:30:"rootcandy_navigation_source_17";s:0:"";s:29:"rootcandy_navigation_source_9";s:0:"";s:29:"rootcandy_navigation_source_6";s:0:"";s:30:"rootcandy_navigation_source_15";s:0:"";s:29:"rootcandy_navigation_source_7";s:0:"";s:29:"rootcandy_navigation_source_3";s:0:"";s:4:"data";a:1:{i:0;s:15:"Support Manager";}s:13:"role-weight-1";s:1:"1";s:13:"role-weight-2";s:1:"2";s:14:"role-weight-14";s:1:"3";s:14:"role-weight-16";s:2:"10";s:14:"role-weight-13";s:1:"4";s:14:"role-weight-18";s:2:"11";s:14:"role-weight-17";s:2:"12";s:13:"role-weight-9";s:1:"5";s:13:"role-weight-6";s:1:"6";s:14:"role-weight-15";s:1:"7";s:13:"role-weight-7";s:1:"8";s:13:"role-weight-3";s:1:"9";}" string(1961) "a:53:{s:11:"toggle_logo";i:0;s:11:"toggle_name";i:1;s:13:"toggle_slogan";i:0;s:14:"toggle_mission";i:1;s:24:"toggle_node_user_picture";i:0;s:27:"toggle_comment_user_picture";i:0;s:13:"toggle_search";i:0;s:14:"toggle_favicon";i:1;s:20:"toggle_primary_links";i:0;s:22:"toggle_secondary_links";i:0;s:12:"default_logo";i:0;s:9:"logo_path";s:49:"sites/default/files/avadis-logo-large.png ";s:11:"logo_upload";s:0:"";s:15:"default_favicon";i:1;s:12:"favicon_path";s:0:"";s:14:"favicon_upload";s:0:"";s:24:"rootcandy_header_display";i:0;s:20:"rootcandy_hide_panel";i:0;s:27:"rootcandy_dashboard_display";i:0;s:24:"rootcandy_dashboard_help";s:4:"left";s:28:"rootcandy_dashboard_messages";s:5:"right";s:35:"rootcandy_dashboard_content_display";i:0;s:26:"rootcandy_navigation_icons";i:0;s:31:"rootcandy_navigation_icons_size";s:2:"24";s:33:"rootcandy_navigation_source_admin";s:29:"_rootcandy_default_navigation";s:29:"rootcandy_navigation_source_1";s:0:"";s:29:"rootcandy_navigation_source_2";s:0:"";s:30:"rootcandy_navigation_source_14";s:0:"";s:30:"rootcandy_navigation_source_16";s:0:"";s:30:"rootcandy_navigation_source_13";s:0:"";s:30:"rootcandy_navigation_source_18";s:0:"";s:30:"rootcandy_navigation_source_17";s:0:"";s:29:"rootcandy_navigation_source_9";s:0:"";s:29:"rootcandy_navigation_source_6";s:0:"";s:30:"rootcandy_navigation_source_15";s:0:"";s:29:"rootcandy_navigation_source_7";s:0:"";s:29:"rootcandy_navigation_source_3";s:0:"";s:33:"rootcandy_navigation_custom_icons";s:0:"";s:22:"rootcandy_help_display";i:0;s:21:"rootcandy_hide_author";i:0;s:4:"data";a:1:{i:0;s:15:"Support Manager";}s:13:"role-weight-1";s:1:"1";s:13:"role-weight-2";s:1:"2";s:14:"role-weight-14";s:1:"3";s:14:"role-weight-16";s:1:"4";s:14:"role-weight-13";s:1:"5";s:14:"role-weight-18";s:1:"6";s:14:"role-weight-17";s:1:"7";s:13:"role-weight-9";s:1:"8";s:13:"role-weight-6";s:1:"9";s:14:"role-weight-15";s:2:"10";s:13:"role-weight-7";s:2:"11";s:13:"role-weight-3";s:2:"12";}" string(4) "i:0;" string(4) "i:0;" string(4) "i:1;" string(11) "s:4:"each";" string(11) "s:4:"each";" string(11) "s:4:"each";" string(11) "s:4:"each";" string(11) "s:4:"each";" string(11) "s:4:"each";" string(11) "s:4:"each";" string(22) "a:1:{i:0;s:5:"title";}" string(6) "a:0:{}" string(6) "a:0:{}" string(6) "a:0:{}" string(22) "a:1:{i:0;s:5:"title";}" string(6) "a:0:{}" string(6) "a:0:{}" string(11) "s:4:"type";" string(11) "s:4:"type";" string(11) "s:4:"type";" string(11) "s:4:"type";" string(11) "s:4:"type";" string(11) "s:4:"type";" string(11) "s:4:"type";" string(30) "a:1:{i:0;s:12:"show_matches";}" string(6) "a:0:{}" string(6) "a:0:{}" string(6) "a:0:{}" string(6) "a:0:{}" string(6) "a:0:{}" string(6) "a:0:{}" string(8) "s:1:"1";" string(8) "s:1:"1";" string(8) "s:1:"1";" string(8) "s:1:"1";" string(8) "s:1:"1";" string(8) "s:1:"1";" string(8) "s:1:"1";" string(8) "s:1:"1";" string(8) "s:1:"1";" string(8) "s:1:"1";" string(250) "a:3:{s:32:"fc839aa0131e164d6f2da95e93629110";s:35:"new_installation_state_view-block_1";s:32:"a2a3687f1ac1886497a0acfebe31b6c5";s:32:"new_product_updates_view-block_1";s:32:"9ad313584d6ebd5179deb6652102068a";s:33:"profile_personal_editable-block_1";}" string(7) "s:0:"";" string(4) "i:0;" string(8) "s:1:"0";" string(46) "s:38:"http://localhost/w3c-markup-validator/";" string(28) "s:20:"w3c_markup_validator";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(7) "s:0:"";" string(33) "s:25:"http://www.strand-ngs.com";" string(10) "s:3:"100";" string(4) "i:1;" string(46) "s:38:"https://www.strand-ngs.com/sitemap.xml";" string(87) "a:5:{i:0;s:3:"ask";i:1;s:4:"bing";i:2;s:6:"google";i:3;s:8:"moreover";i:4;s:5:"yahoo";}" string(12) "s:5:"86400";" string(13) "i:1591074002;" string(4) "i:1;" string(15) "s:8:"31449600";" string(10) "s:3:"1.0";" string(13) "i:1591074002;" string(20) "s:12:"Y-m-d\TH:i\Z";" string(4) "i:0;" string(4) "i:0;" string(10) "s:3:"300";" string(22) "s:14:"crm-xmlsitemap";" string(4) "i:1;" string(4) "b:0;" string(4) "b:1;" string(52) "a:2:{s:6:"status";s:1:"0";s:8:"priority";s:3:"0.5";}" string(52) "a:2:{s:6:"status";s:1:"0";s:8:"priority";s:3:"0.5";}" string(52) "a:2:{s:6:"status";s:1:"0";s:8:"priority";s:3:"0.5";}" string(52) "a:2:{s:6:"status";s:1:"1";s:8:"priority";s:3:"0.5";}" string(52) "a:2:{s:6:"status";s:1:"0";s:8:"priority";s:3:"0.5";}" string(52) "a:2:{s:6:"status";s:1:"1";s:8:"priority";s:3:"0.5";}" string(52) "a:2:{s:6:"status";s:1:"0";s:8:"priority";s:3:"0.5";}" string(52) "a:2:{s:6:"status";s:1:"0";s:8:"priority";s:3:"0.5";}" string(52) "a:2:{s:6:"status";s:1:"1";s:8:"priority";s:3:"0.5";}" string(4) "i:1;" string(52) "a:2:{s:6:"status";s:1:"0";s:8:"priority";s:3:"0.5";}" string(15) "s:8:"comments";" string(20) "s:12:"nodecomments";" string(676) "a:6:{s:14:"rootcandy_dark";a:2:{s:9:"menu_item";s:15:"theme_menu_item";s:14:"menu_item_link";s:24:"rootcandy_menu_item_link";}s:8:"newtheme";a:2:{s:9:"menu_item";s:15:"theme_menu_item";s:14:"menu_item_link";s:18:"zen_menu_item_link";}s:9:"sitetheme";a:2:{s:9:"menu_item";s:15:"theme_menu_item";s:14:"menu_item_link";s:18:"zen_menu_item_link";}s:8:"minnelli";a:2:{s:9:"menu_item";s:15:"theme_menu_item";s:14:"menu_item_link";s:20:"theme_menu_item_link";}s:10:"bluemarine";a:2:{s:9:"menu_item";s:15:"theme_menu_item";s:14:"menu_item_link";s:20:"theme_menu_item_link";}s:9:"strandngs";a:2:{s:9:"menu_item";s:15:"theme_menu_item";s:14:"menu_item_link";s:18:"zen_menu_item_link";}}" string(52) "a:2:{s:6:"status";s:1:"0";s:8:"priority";s:3:"0.5";}" string(8) "s:1:"0";" string(4) "i:1;" string(8) "s:1:"0";" string(9) "s:2:"50";" string(8) "s:1:"3";" string(4) "i:0;" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"1";" string(8) "s:1:"0";" string(11) "s:4:"none";" string(4) "i:0;" string(78) "s:70:"kondo@agilent.com, kohei_ando@agilent.com, yoshiyuki_ishii@agilent.com";" string(11) "s:4:"only";" string(41) "a:2:{i:0;s:6:"status";i:1;s:7:"promote";}" string(8) "s:1:"0";" string(4) "i:0;" string(4) "i:1;" string(4) "i:0;" string(7) "s:0:"";" string(3323) "a:20:{s:11:"toggle_logo";i:1;s:11:"toggle_name";i:0;s:13:"toggle_slogan";i:0;s:14:"toggle_mission";i:1;s:24:"toggle_node_user_picture";i:0;s:27:"toggle_comment_user_picture";i:0;s:13:"toggle_search";i:0;s:14:"toggle_favicon";i:1;s:20:"toggle_primary_links";i:0;s:22:"toggle_secondary_links";i:0;s:12:"default_logo";i:0;s:9:"logo_path";s:43:"sites/default/files/strand_logo.jpg ";s:11:"logo_upload";s:0:"";s:15:"default_favicon";i:1;s:12:"favicon_path";s:0:"";s:14:"favicon_upload";s:0:"";s:6:"scheme";s:39:"#0072b9,#027ac6,#2385c2,#5ab5ee,#494949";s:7:"palette";a:5:{s:4:"base";s:7:"#0072b9";s:4:"link";s:7:"#027ac6";s:3:"top";s:7:"#2385c2";s:6:"bottom";s:7:"#5ab5ee";s:4:"text";s:7:"#494949";}s:5:"theme";s:8:"minnelli";s:4:"info";a:10:{s:7:"schemes";a:16:{s:39:"#0072b9,#027ac6,#2385c2,#5ab5ee,#494949";s:21:"Blue Lagoon (Default)";s:39:"#464849,#2f416f,#2a2b2d,#5d6779,#494949";s:3:"Ash";s:39:"#55c0e2,#000000,#085360,#007e94,#696969";s:10:"Aquamarine";s:39:"#d5b048,#6c420e,#331900,#971702,#494949";s:17:"Belgian Chocolate";s:39:"#3f3f3f,#336699,#6598cb,#6598cb,#000000";s:10:"Bluemarine";s:39:"#d0cb9a,#917803,#efde01,#e6fb2d,#494949";s:12:"Citrus Blast";s:39:"#0f005c,#434f8c,#4d91ff,#1a1575,#000000";s:8:"Cold Day";s:39:"#c9c497,#0c7a00,#03961e,#7be000,#494949";s:9:"Greenbeam";s:39:"#ffe23d,#a9290a,#fc6d1d,#a30f42,#494949";s:11:"Mediterrano";s:39:"#788597,#3f728d,#a9adbc,#d4d4d4,#707070";s:7:"Mercury";s:39:"#5b5fa9,#5b5faa,#0a2352,#9fa8d5,#494949";s:9:"Nocturnal";s:39:"#7db323,#6a9915,#b5d52a,#7db323,#191a19";s:6:"Olivia";s:39:"#12020b,#1b1a13,#f391c6,#f41063,#898080";s:12:"Pink Plastic";s:39:"#b7a0ba,#c70000,#a1443a,#f21107,#515d52";s:12:"Shiny Tomato";s:39:"#18583d,#1b5f42,#34775a,#52bf90,#2d2d2d";s:8:"Teal Top";s:0:"";s:6:"Custom";}s:4:"copy";a:4:{i:0;s:28:"../images/menu-collapsed.gif";i:1;s:32:"../images/menu-collapsed-rtl.gif";i:2;s:27:"../images/menu-expanded.gif";i:3;s:23:"../images/menu-leaf.gif";}s:3:"css";a:1:{i:0;s:12:"../style.css";}s:8:"gradient";a:4:{i:0;i:0;i:1;i:37;i:2;i:760;i:3;i:121;}s:4:"fill";a:2:{s:4:"base";a:4:{i:0;i:0;i:1;i:0;i:2;i:760;i:3;i:568;}s:4:"link";a:4:{i:0;i:107;i:1;i:533;i:2;i:41;i:3;i:23;}}s:6:"slices";a:13:{s:18:"../images/body.png";a:4:{i:0;i:0;i:1;i:37;i:2;i:1;i:3;i:280;}s:20:"../images/bg-bar.png";a:4:{i:0;i:202;i:1;i:530;i:2;i:76;i:3;i:14;}s:26:"../images/bg-bar-white.png";a:4:{i:0;i:202;i:1;i:506;i:2;i:76;i:3;i:14;}s:20:"../images/bg-tab.png";a:4:{i:0;i:107;i:1;i:533;i:2;i:41;i:3;i:23;}s:27:"../images/bg-navigation.png";a:4:{i:0;i:0;i:1;i:0;i:2;i:7;i:3;i:37;}s:29:"../images/bg-content-left.png";a:4:{i:0;i:40;i:1;i:117;i:2;i:50;i:3;i:352;}s:30:"../images/bg-content-right.png";a:4:{i:0;i:510;i:1;i:117;i:2;i:50;i:3;i:352;}s:24:"../images/bg-content.png";a:4:{i:0;i:299;i:1;i:117;i:2;i:7;i:3;i:200;}s:32:"../images/bg-navigation-item.png";a:4:{i:0;i:32;i:1;i:37;i:2;i:17;i:3;i:12;}s:38:"../images/bg-navigation-item-hover.png";a:4:{i:0;i:54;i:1;i:37;i:2;i:17;i:3;i:12;}s:28:"../images/gradient-inner.png";a:4:{i:0;i:646;i:1;i:307;i:2;i:112;i:3;i:42;}s:8:"logo.png";a:4:{i:0;i:622;i:1;i:51;i:2;i:64;i:3;i:73;}s:14:"screenshot.png";a:4:{i:0;i:0;i:1;i:37;i:2;i:400;i:3;i:240;}}s:12:"blend_target";s:7:"#ffffff";s:13:"preview_image";s:17:"color/preview.png";s:11:"preview_css";s:20:"../color/preview.css";s:10:"base_image";s:14:"color/base.png";}}" string(4) "i:0;" string(4) "i:1;" string(8) "s:1:"5";" string(8) "s:1:"3";" string(466) "a:21:{s:7:"profile";s:7:"profile";s:7:"account";i:0;s:7:"comment";i:0;s:5:"event";i:0;s:11:"interaction";i:0;s:8:"internal";i:0;s:19:"non_searchable_page";i:0;s:11:"opportunity";i:0;s:20:"support_organization";i:0;s:4:"page";i:0;s:7:"product";i:0;s:12:"product_page";i:0;s:18:"product_quote_form";i:0;s:6:"region";i:0;s:16:"software_product";i:0;s:5:"story";i:0;s:15:"support_product";i:0;s:6:"survey";i:0;s:13:"supportticket";i:0;s:5:"video";i:0;s:7:"webform";i:0;}" string(4220) "a:28:{s:5:"title";a:3:{s:6:"region";s:4:"main";s:6:"weight";s:1:"7";s:6:"hidden";i:1;}s:4:"menu";a:6:{s:6:"region";s:6:"footer";s:6:"weight";s:1:"9";s:12:"has_required";b:0;s:5:"title";s:13:"Menu settings";s:9:"collapsed";i:1;s:6:"hidden";i:0;}s:20:"revision_information";a:6:{s:6:"region";s:6:"footer";s:6:"weight";s:1:"7";s:12:"has_required";b:0;s:5:"title";s:20:"Revision information";s:9:"collapsed";i:1;s:6:"hidden";i:0;}s:16:"comment_settings";a:6:{s:6:"region";s:6:"footer";s:6:"weight";s:1:"3";s:12:"has_required";b:0;s:5:"title";s:16:"Comment settings";s:9:"collapsed";i:1;s:6:"hidden";i:0;}s:4:"path";a:6:{s:6:"region";s:6:"footer";s:6:"weight";s:1:"4";s:12:"has_required";b:0;s:5:"title";s:17:"URL path settings";s:9:"collapsed";i:1;s:6:"hidden";i:0;}s:7:"options";a:6:{s:6:"region";s:6:"footer";s:6:"weight";s:1:"5";s:12:"has_required";b:0;s:5:"title";s:18:"Publishing options";s:9:"collapsed";i:1;s:6:"hidden";i:0;}s:6:"author";a:6:{s:6:"region";s:6:"footer";s:6:"weight";s:1:"8";s:12:"has_required";b:0;s:5:"title";s:21:"Authoring information";s:9:"collapsed";i:1;s:6:"hidden";i:0;}s:7:"buttons";a:5:{s:6:"region";s:6:"footer";s:6:"weight";s:2:"10";s:12:"has_required";b:0;s:5:"title";N;s:6:"hidden";i:0;}s:11:"attachments";a:6:{s:6:"region";s:4:"main";s:6:"weight";s:1:"9";s:12:"has_required";b:0;s:5:"title";s:16:"File attachments";s:9:"collapsed";i:1;s:6:"hidden";i:0;}s:16:"field_start_time";a:5:{s:6:"region";s:4:"main";s:6:"weight";s:1:"2";s:12:"has_required";b:0;s:5:"title";s:16:"Time information";s:6:"hidden";i:1;}s:16:"field_reviewedby";a:5:{s:6:"region";s:5:"right";s:6:"weight";s:1:"3";s:12:"has_required";b:0;s:5:"title";s:11:"Reviewed by";s:6:"hidden";i:0;}s:10:"field_mode";a:4:{s:6:"region";s:5:"right";s:6:"weight";s:1:"0";s:12:"has_required";b:1;s:5:"title";s:4:"Mode";}s:10:"field_mood";a:4:{s:6:"region";s:5:"right";s:6:"weight";s:1:"1";s:12:"has_required";b:1;s:5:"title";s:4:"Mood";}s:28:"field_interaction_ticket_vid";a:5:{s:6:"region";s:4:"main";s:6:"weight";s:1:"8";s:12:"has_required";b:0;s:5:"title";s:17:"Ticket Version Id";s:6:"hidden";i:1;}s:17:"field_substantive";a:5:{s:6:"region";s:5:"right";s:6:"weight";s:1:"4";s:12:"has_required";b:0;s:5:"title";s:21:"Substantial Response ";s:6:"hidden";i:0;}s:19:"field_show_customer";a:5:{s:6:"region";s:5:"right";s:6:"weight";s:1:"6";s:12:"has_required";b:0;s:5:"title";s:28:"Show Interaction to customer";s:6:"hidden";i:0;}s:21:"field_self_assessment";a:4:{s:6:"region";s:5:"right";s:6:"weight";s:1:"2";s:12:"has_required";b:1;s:5:"title";s:15:"Self assessment";}s:18:"field_mark_deleted";a:5:{s:6:"region";s:5:"right";s:6:"weight";s:1:"5";s:12:"has_required";b:0;s:5:"title";s:15:"Mark as deleted";s:6:"hidden";i:0;}s:26:"field_interaction_customer";a:4:{s:6:"region";s:4:"main";s:6:"weight";s:1:"3";s:12:"has_required";b:1;s:5:"title";s:20:"Interaction:Customer";}s:25:"field_interaction_support";a:4:{s:6:"region";s:4:"main";s:6:"weight";s:1:"6";s:12:"has_required";b:1;s:5:"title";s:16:"Support Response";}s:6:"domain";a:5:{s:6:"region";s:6:"footer";s:6:"weight";s:1:"0";s:12:"has_required";b:1;s:5:"title";s:21:"Domain access options";s:9:"collapsed";i:1;}i:0;a:5:{s:6:"region";s:6:"footer";s:6:"weight";s:1:"6";s:12:"has_required";b:0;s:5:"title";N;s:6:"hidden";i:0;}s:10:"xmlsitemap";a:6:{s:6:"region";s:6:"footer";s:6:"weight";s:1:"2";s:12:"has_required";b:0;s:5:"title";s:11:"XML sitemap";s:9:"collapsed";i:1;s:6:"hidden";i:0;}s:9:"nodewords";a:6:{s:6:"region";s:6:"footer";s:6:"weight";s:1:"1";s:12:"has_required";b:0;s:5:"title";s:9:"Meta tags";s:9:"collapsed";i:1;s:6:"hidden";i:0;}s:30:"field_interaction_duration_ngs";a:5:{s:6:"region";s:4:"main";s:6:"weight";s:1:"0";s:12:"has_required";b:0;s:5:"title";s:25:"Time spent on Interaction";s:6:"hidden";i:1;}s:4:"flag";a:6:{s:6:"region";s:4:"main";s:6:"weight";s:1:"4";s:12:"has_required";b:0;s:5:"title";s:5:"Flags";s:9:"collapsed";i:0;s:6:"hidden";i:0;}s:17:"field_int_in_time";a:5:{s:6:"region";s:4:"main";s:6:"weight";s:1:"1";s:12:"has_required";b:1;s:5:"title";s:19:"Interaction in-time";s:6:"hidden";i:0;}s:18:"field_int_out_time";a:5:{s:6:"region";s:4:"main";s:6:"weight";s:1:"5";s:12:"has_required";b:1;s:5:"title";s:20:"Interaction out-time";s:6:"hidden";i:0;}}" string(4) "b:0;" string(8) "s:1:"1";" string(4) "i:1;" string(4) "i:0;" string(4) "i:1;" string(45) "s:37:"form-8fc3bad15a59b0068b30cca602f57b82";" string(15) "s:8:"disabled";" string(8) "s:1:"0";" string(4) "i:0;" string(8) "s:1:"0";" string(8) "s:1:"3";" string(4) "i:1;" string(6) "a:0:{}" string(7) "s:0:"";" string(4) "i:0;" string(6) "a:0:{}" string(8) "s:1:"0";" string(7) "s:0:"";" string(4) "i:0;" string(4) "i:0;" string(4) "i:0;" string(7) "s:0:"";" string(11) "s:4:"show";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(11) "s:4:"both";" string(11) "s:4:"show";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(11) "s:4:"both";" string(7) "s:0:"";" string(4) "i:1;" string(31) "s:23:"flowplayer3_mediaplayer";" string(31) "s:23:"flowplayer3_mediaplayer";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(31) "s:23:"flowplayer3_mediaplayer";" string(31) "s:23:"flowplayer3_mediaplayer";" string(31) "s:23:"flowplayer3_mediaplayer";" string(11) "s:4:"here";" string(4) "i:1;" string(36) "s:28:"swf flv xml mp3 jpg jpeg png";" string(11) "s:4:"each";" string(4) "i:1;" string(8) "s:1:"0";" string(8) "s:1:"3";" string(4) "i:1;" string(6) "a:0:{}" string(7) "s:0:"";" string(4) "i:0;" string(8) "s:1:"2";" string(4) "i:1;" string(45) "s:37:"form-1a90b4f9eb1a2b4396fa1b0cd0bfebf0";" string(4) "i:0;" string(4) "i:0;" string(4) "i:0;" string(8) "s:1:"1";" string(21) "s:13:"questions_top";" string(9) "s:2:"ul";" string(4) "i:0;" string(7) "s:0:"";" string(7) "s:0:"";" string(11) "s:4:"long";" string(4) "i:0;" string(4) "i:1;" string(4) "i:0;" string(4) "i:0;" string(7) "s:0:"";" string(4) "i:0;" string(11) "s:4:"DESC";" string(8) "s:1:"0";" string(6) "a:0:{}" string(11) "s:4:"type";" string(11) "s:4:"each";" string(6) "a:0:{}" string(8) "s:1:"4";" string(8) "s:1:"1";" string(9) "s:2:"50";" string(8) "s:1:"3";" string(4) "i:0;" string(8) "s:1:"1";" string(8) "s:1:"1";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(11) "s:4:"none";" string(4) "i:0;" string(7) "s:0:"";" string(15) "s:8:"comments";" string(20) "s:12:"nodecomments";" string(8) "s:1:"0";" string(23) "a:1:{i:0;s:6:"enable";}" string(284) "a:11:{s:5:"title";s:2:"-5";s:20:"revision_information";s:2:"20";s:6:"author";s:2:"20";s:7:"options";s:2:"25";s:16:"comment_settings";s:2:"30";s:4:"menu";s:2:"-2";s:4:"path";s:2:"30";s:11:"attachments";s:2:"30";s:6:"domain";s:1:"1";s:10:"xmlsitemap";s:2:"30";s:9:"nodewords";s:2:"10";}" string(7) "s:0:"";" string(16) "s:9:"playlists";" string(6) "a:0:{}" string(8) "s:1:"1";" string(7) "s:0:"";" string(4) "i:0;" string(4) "i:1;" string(4) "i:0;" string(7) "s:0:"";" string(52) "a:2:{s:6:"status";s:1:"0";s:8:"priority";s:3:"0.5";}" string(8) "s:1:"0";" string(6) "a:0:{}" string(7) "s:0:"";" string(11) "s:4:"both";" string(34) "s:26:"Frequently Asked Questions";" string(212) "s:203:" ";" string(8) "s:1:"2";" string(4944) "a:32:{s:5:"title";a:4:{s:6:"region";s:4:"main";s:6:"weight";s:1:"0";s:12:"has_required";b:1;s:5:"title";s:4:"Name";}s:4:"menu";a:6:{s:6:"region";s:6:"footer";s:6:"weight";s:1:"7";s:12:"has_required";b:0;s:5:"title";s:13:"Menu settings";s:9:"collapsed";i:1;s:6:"hidden";i:0;}s:20:"revision_information";a:6:{s:6:"region";s:6:"footer";s:6:"weight";s:1:"5";s:12:"has_required";b:0;s:5:"title";s:20:"Revision information";s:9:"collapsed";i:1;s:6:"hidden";i:0;}s:16:"comment_settings";a:6:{s:6:"region";s:6:"footer";s:6:"weight";s:1:"2";s:12:"has_required";b:0;s:5:"title";s:16:"Comment settings";s:9:"collapsed";i:1;s:6:"hidden";i:0;}s:4:"path";a:6:{s:6:"region";s:6:"footer";s:6:"weight";s:1:"3";s:12:"has_required";b:0;s:5:"title";s:17:"URL path settings";s:9:"collapsed";i:1;s:6:"hidden";i:0;}s:7:"options";a:6:{s:6:"region";s:6:"footer";s:6:"weight";s:1:"4";s:12:"has_required";b:0;s:5:"title";s:18:"Publishing options";s:9:"collapsed";i:1;s:6:"hidden";i:0;}s:6:"author";a:6:{s:6:"region";s:6:"footer";s:6:"weight";s:1:"6";s:12:"has_required";b:0;s:5:"title";s:21:"Authoring information";s:9:"collapsed";i:1;s:6:"hidden";i:0;}s:7:"buttons";a:5:{s:6:"region";s:6:"footer";s:6:"weight";s:1:"8";s:12:"has_required";b:0;s:5:"title";N;s:6:"hidden";i:0;}s:11:"attachments";a:6:{s:6:"region";s:4:"main";s:6:"weight";s:2:"17";s:12:"has_required";b:0;s:5:"title";s:16:"File attachments";s:9:"collapsed";i:1;s:6:"hidden";i:0;}s:19:"field_account_phone";a:5:{s:6:"region";s:4:"main";s:6:"weight";s:1:"2";s:12:"has_required";b:0;s:5:"title";s:5:"Phone";s:6:"hidden";i:0;}s:22:"field_account_academic";a:5:{s:6:"region";s:4:"main";s:6:"weight";s:1:"3";s:12:"has_required";b:0;s:5:"title";s:11:"Is academic";s:6:"hidden";i:0;}s:20:"field_account_vendor";a:4:{s:6:"region";s:4:"main";s:6:"weight";s:1:"4";s:12:"has_required";b:1;s:5:"title";s:6:"Vendor";}s:26:"field_account_primary_user";a:5:{s:6:"region";s:5:"right";s:6:"weight";s:1:"8";s:12:"has_required";b:0;s:5:"title";s:12:"Primary User";s:6:"hidden";i:0;}s:21:"field_account_onwatch";a:4:{s:6:"region";s:4:"main";s:6:"weight";s:1:"6";s:12:"has_required";b:1;s:5:"title";s:8:"On watch";}s:26:"field_account_onwatch_till";a:5:{s:6:"region";s:4:"main";s:6:"weight";s:1:"8";s:12:"has_required";b:0;s:5:"title";s:17:"On watch End Date";s:6:"hidden";i:0;}s:21:"field_account_address";a:5:{s:6:"region";s:4:"main";s:6:"weight";s:1:"1";s:12:"has_required";b:0;s:5:"title";s:7:"Address";s:6:"hidden";i:0;}s:16:"field_account_to";a:5:{s:6:"region";s:5:"right";s:6:"weight";s:1:"0";s:12:"has_required";b:0;s:5:"title";s:17:"Sales To Contacts";s:6:"hidden";i:0;}s:22:"field_account_to_roles";a:5:{s:6:"region";s:5:"right";s:6:"weight";s:1:"1";s:12:"has_required";b:0;s:5:"title";s:14:"Sales To Roles";s:6:"hidden";i:0;}s:16:"field_account_cc";a:5:{s:6:"region";s:5:"right";s:6:"weight";s:1:"2";s:12:"has_required";b:0;s:5:"title";s:17:"Sales CC contacts";s:6:"hidden";i:0;}s:22:"field_account_cc_roles";a:5:{s:6:"region";s:5:"right";s:6:"weight";s:1:"3";s:12:"has_required";b:0;s:5:"title";s:14:"Sales CC roles";s:6:"hidden";i:0;}s:21:"field_account_support";a:5:{s:6:"region";s:5:"right";s:6:"weight";s:1:"4";s:12:"has_required";b:0;s:5:"title";s:16:"Support To Users";s:6:"hidden";i:0;}s:27:"field_account_support_roles";a:5:{s:6:"region";s:5:"right";s:6:"weight";s:1:"7";s:12:"has_required";b:0;s:5:"title";s:16:"Support To Roles";s:6:"hidden";i:0;}s:30:"field_account_support_cc_users";a:5:{s:6:"region";s:5:"right";s:6:"weight";s:1:"6";s:12:"has_required";b:0;s:5:"title";s:18:"Support CC Contact";s:6:"hidden";i:0;}s:30:"field_account_support_cc_roles";a:5:{s:6:"region";s:5:"right";s:6:"weight";s:1:"5";s:12:"has_required";b:0;s:5:"title";s:16:"Support CC Roles";s:6:"hidden";i:0;}s:6:"domain";a:5:{s:6:"region";s:4:"main";s:6:"weight";s:2:"18";s:12:"has_required";b:1;s:5:"title";s:21:"Domain access options";s:9:"collapsed";i:1;}i:0;a:5:{s:6:"region";s:6:"footer";s:6:"weight";s:1:"7";s:12:"has_required";b:0;s:5:"title";N;s:6:"hidden";i:0;}s:10:"xmlsitemap";a:6:{s:6:"region";s:6:"footer";s:6:"weight";s:1:"1";s:12:"has_required";b:0;s:5:"title";s:11:"XML sitemap";s:9:"collapsed";i:1;s:6:"hidden";i:0;}s:8:"group_to";a:6:{s:6:"region";s:5:"right";s:6:"weight";s:1:"9";s:12:"has_required";b:0;s:5:"title";s:8:"Sales To";s:9:"collapsed";i:1;s:6:"hidden";i:0;}s:8:"group_cc";a:6:{s:6:"region";s:5:"right";s:6:"weight";s:2:"10";s:12:"has_required";b:0;s:5:"title";s:8:"Sales CC";s:9:"collapsed";i:1;s:6:"hidden";i:0;}s:21:"group_account_support";a:6:{s:6:"region";s:5:"right";s:6:"weight";s:2:"11";s:12:"has_required";b:0;s:5:"title";s:10:"Support To";s:9:"collapsed";i:1;s:6:"hidden";i:0;}s:24:"group_account_support_cc";a:5:{s:6:"region";s:5:"right";s:6:"weight";s:2:"12";s:12:"has_required";b:0;s:5:"title";s:10:"Support cc";s:6:"hidden";i:0;}s:9:"nodewords";a:6:{s:6:"region";s:6:"footer";s:6:"weight";s:1:"0";s:12:"has_required";b:0;s:5:"title";s:9:"Meta tags";s:9:"collapsed";i:1;s:6:"hidden";i:0;}}" string(26) "s:18:"swfobject2_replace";" string(4) "i:1;" string(157) "s:148:"You are missing some Flash content that should appear here! Perhaps your browser cannot display it, or maybe it did not initialize correctly.
";" string(8) "s:1:"7";" string(4) "i:1;" string(4) "i:1;" string(4) "i:0;" string(4) "i:1;" string(14) "s:7:"#FFFFFF";" string(15) "s:8:"autohigh";" string(14) "s:7:"showall";" string(13) "s:6:"opaque";" string(8) "s:1:"l";" string(9) "s:2:"tl";" string(14) "s:7:"default";" string(18) "s:10:"sameDomain";" string(11) "s:4:"type";" string(15) "s:8:"disabled";" string(11) "s:4:"show";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(11) "s:4:"both";" string(11) "s:4:"show";" string(7) "s:0:"";" string(7) "s:0:"";" string(2) "N;" string(4) "i:0;" string(6) "a:0:{}" string(8) "s:1:"0";" string(8) "s:1:"1";" string(4) "i:1;" string(5154) "a:34:{s:5:"title";a:4:{s:6:"region";s:4:"main";s:6:"weight";s:1:"0";s:12:"has_required";b:1;s:5:"title";s:5:"Title";}s:4:"menu";a:6:{s:6:"region";s:6:"footer";s:6:"weight";s:1:"7";s:12:"has_required";b:0;s:5:"title";s:13:"Menu settings";s:9:"collapsed";i:1;s:6:"hidden";i:1;}s:20:"revision_information";a:6:{s:6:"region";s:4:"main";s:6:"weight";s:1:"6";s:12:"has_required";b:0;s:5:"title";s:20:"Revision information";s:9:"collapsed";i:0;s:6:"hidden";i:0;}s:16:"comment_settings";a:6:{s:6:"region";s:6:"footer";s:6:"weight";s:1:"3";s:12:"has_required";b:0;s:5:"title";s:16:"Comment settings";s:9:"collapsed";i:1;s:6:"hidden";i:1;}s:4:"path";a:6:{s:6:"region";s:6:"footer";s:6:"weight";s:1:"4";s:12:"has_required";b:0;s:5:"title";s:17:"URL path settings";s:9:"collapsed";i:1;s:6:"hidden";i:1;}s:7:"options";a:6:{s:6:"region";s:6:"footer";s:6:"weight";s:1:"5";s:12:"has_required";b:0;s:5:"title";s:18:"Publishing options";s:9:"collapsed";i:1;s:6:"hidden";i:1;}s:6:"author";a:6:{s:6:"region";s:6:"footer";s:6:"weight";s:1:"6";s:12:"has_required";b:0;s:5:"title";s:21:"Authoring information";s:9:"collapsed";i:1;s:6:"hidden";i:1;}s:7:"buttons";a:5:{s:6:"region";s:6:"footer";s:6:"weight";s:1:"8";s:12:"has_required";b:0;s:5:"title";N;s:6:"hidden";i:0;}s:8:"taxonomy";a:5:{s:6:"region";s:4:"main";s:6:"weight";s:1:"1";s:12:"has_required";b:1;s:5:"title";s:14:"Categorisation";s:9:"collapsed";i:0;}s:11:"attachments";a:6:{s:6:"region";s:4:"main";s:6:"weight";s:1:"5";s:12:"has_required";b:0;s:5:"title";s:16:"File attachments";s:9:"collapsed";i:1;s:6:"hidden";i:0;}s:14:"field_customer";a:4:{s:6:"region";s:4:"main";s:6:"weight";s:1:"7";s:12:"has_required";b:1;s:5:"title";s:13:"Customer Name";}s:24:"field_ticket_assigned_to";a:5:{s:6:"region";s:5:"right";s:6:"weight";s:1:"1";s:12:"has_required";b:0;s:5:"title";s:11:"Assigned To";s:6:"hidden";i:0;}s:19:"field_ticket_serial";a:5:{s:6:"region";s:4:"main";s:6:"weight";s:1:"8";s:12:"has_required";b:0;s:5:"title";s:13:"Ticket Serial";s:6:"hidden";i:1;}s:15:"field_ticket_id";a:5:{s:6:"region";s:4:"main";s:6:"weight";s:1:"9";s:12:"has_required";b:0;s:5:"title";s:9:"Ticket ID";s:6:"hidden";i:1;}s:25:"field_user_contact_source";a:4:{s:6:"region";s:4:"main";s:6:"weight";s:1:"2";s:12:"has_required";b:1;s:5:"title";s:6:"Source";}s:18:"field_ticket_court";a:4:{s:6:"region";s:4:"main";s:6:"weight";s:1:"3";s:12:"has_required";b:1;s:5:"title";s:5:"Court";}s:18:"field_ticketstatus";a:4:{s:6:"region";s:4:"main";s:6:"weight";s:1:"4";s:12:"has_required";b:1;s:5:"title";s:6:"Status";}s:23:"field_productmanagement";a:5:{s:6:"region";s:5:"right";s:6:"weight";s:1:"7";s:12:"has_required";b:0;s:5:"title";s:30:"Reviewed by Product Management";s:6:"hidden";i:0;}s:19:"field_ignore_ticket";a:5:{s:6:"region";s:5:"right";s:6:"weight";s:1:"5";s:12:"has_required";b:0;s:5:"title";s:19:"Ignore from Reports";s:6:"hidden";i:0;}s:17:"field_handingover";a:5:{s:6:"region";s:5:"right";s:6:"weight";s:1:"4";s:12:"has_required";b:0;s:5:"title";s:8:"Handover";s:6:"hidden";i:0;}s:13:"field_renewal";a:5:{s:6:"region";s:5:"right";s:6:"weight";s:1:"6";s:12:"has_required";b:0;s:5:"title";s:20:"Critical for Renewal";s:6:"hidden";i:0;}s:15:"field_user_leed";a:5:{s:6:"region";s:5:"right";s:6:"weight";s:1:"3";s:12:"has_required";b:0;s:5:"title";s:4:"Lead";s:6:"hidden";i:0;}s:15:"field_timestamp";a:5:{s:6:"region";s:4:"main";s:6:"weight";s:2:"10";s:12:"has_required";b:0;s:5:"title";s:21:"Revision Time(Ticket)";s:6:"hidden";i:0;}s:27:"field_total_escalation_time";a:5:{s:6:"region";s:4:"main";s:6:"weight";s:2:"11";s:12:"has_required";b:0;s:5:"title";s:19:"Net Escalation time";s:6:"hidden";i:1;}s:6:"domain";a:5:{s:6:"region";s:6:"footer";s:6:"weight";s:2:"11";s:12:"has_required";b:1;s:5:"title";s:21:"Domain access options";s:9:"collapsed";i:1;}i:0;a:5:{s:6:"region";s:6:"footer";s:6:"weight";s:1:"2";s:12:"has_required";b:0;s:5:"title";N;s:6:"hidden";i:0;}s:13:"customer-info";a:6:{s:6:"region";s:5:"right";s:6:"weight";s:1:"0";s:12:"has_required";b:0;s:5:"title";s:20:"Customer Information";s:9:"collapsed";i:0;s:6:"hidden";i:0;}s:10:"xmlsitemap";a:6:{s:6:"region";s:6:"footer";s:6:"weight";s:1:"0";s:12:"has_required";b:0;s:5:"title";s:11:"XML sitemap";s:9:"collapsed";i:1;s:6:"hidden";i:1;}s:9:"nodewords";a:6:{s:6:"region";s:6:"footer";s:6:"weight";s:1:"1";s:12:"has_required";b:0;s:5:"title";s:9:"Meta tags";s:9:"collapsed";i:1;s:6:"hidden";i:1;}s:23:"field_ticket_basil_link";a:5:{s:6:"region";s:4:"main";s:6:"weight";s:2:"12";s:12:"has_required";b:0;s:5:"title";s:10:"Basil Link";s:6:"hidden";i:0;}s:26:"field_ticket_rnd_escalated";a:5:{s:6:"region";s:4:"main";s:6:"weight";s:2:"13";s:12:"has_required";b:0;s:5:"title";s:13:"RnD Escalated";s:6:"hidden";i:1;}s:25:"field_ticket_pm_escalated";a:5:{s:6:"region";s:4:"main";s:6:"weight";s:2:"14";s:12:"has_required";b:0;s:5:"title";s:12:"PM Escalated";s:6:"hidden";i:1;}s:20:"field_ticket_orderid";a:5:{s:6:"region";s:4:"main";s:6:"weight";s:2:"15";s:12:"has_required";b:0;s:5:"title";s:8:"Order ID";s:6:"hidden";i:0;}s:22:"field_rnd_ticket_owner";a:5:{s:6:"region";s:5:"right";s:6:"weight";s:1:"2";s:12:"has_required";b:0;s:5:"title";s:13:"R&D Owner";s:6:"hidden";i:0;}}" string(3669) "a:24:{s:5:"title";a:3:{s:6:"region";s:4:"main";s:6:"weight";s:1:"8";s:6:"hidden";i:0;}s:4:"menu";a:6:{s:6:"region";s:6:"footer";s:6:"weight";s:1:"9";s:12:"has_required";b:0;s:5:"title";s:13:"Menu settings";s:9:"collapsed";i:1;s:6:"hidden";i:0;}s:20:"revision_information";a:6:{s:6:"region";s:6:"footer";s:6:"weight";s:1:"7";s:12:"has_required";b:0;s:5:"title";s:20:"Revision information";s:9:"collapsed";i:1;s:6:"hidden";i:0;}s:16:"comment_settings";a:6:{s:6:"region";s:6:"footer";s:6:"weight";s:1:"4";s:12:"has_required";b:0;s:5:"title";s:16:"Comment settings";s:9:"collapsed";i:1;s:6:"hidden";i:0;}s:4:"path";a:6:{s:6:"region";s:6:"footer";s:6:"weight";s:1:"5";s:12:"has_required";b:0;s:5:"title";s:17:"URL path settings";s:9:"collapsed";i:1;s:6:"hidden";i:0;}s:7:"options";a:6:{s:6:"region";s:6:"footer";s:6:"weight";s:1:"6";s:12:"has_required";b:0;s:5:"title";s:18:"Publishing options";s:9:"collapsed";i:1;s:6:"hidden";i:0;}s:6:"author";a:6:{s:6:"region";s:6:"footer";s:6:"weight";s:1:"8";s:12:"has_required";b:0;s:5:"title";s:21:"Authoring information";s:9:"collapsed";i:1;s:6:"hidden";i:0;}s:7:"buttons";a:5:{s:6:"region";s:6:"footer";s:6:"weight";s:2:"10";s:12:"has_required";b:0;s:5:"title";N;s:6:"hidden";i:0;}s:11:"attachments";a:6:{s:6:"region";s:4:"main";s:6:"weight";s:2:"10";s:12:"has_required";b:0;s:5:"title";s:16:"File attachments";s:9:"collapsed";i:1;s:6:"hidden";i:0;}s:21:"field_profile_account";a:4:{s:6:"region";s:4:"main";s:6:"weight";s:1:"0";s:12:"has_required";b:1;s:5:"title";s:7:"Account";}s:23:"field_profile_validated";a:4:{s:6:"region";s:5:"right";s:6:"weight";s:1:"1";s:12:"has_required";b:1;s:5:"title";s:12:"Is Validated";}s:23:"field_profile_beta_user";a:5:{s:6:"region";s:5:"right";s:6:"weight";s:1:"0";s:12:"has_required";b:0;s:5:"title";s:9:"Beta User";s:6:"hidden";i:0;}s:20:"field_profile_status";a:5:{s:6:"region";s:4:"main";s:6:"weight";s:1:"3";s:12:"has_required";b:0;s:5:"title";s:6:"Status";s:6:"hidden";i:0;}s:28:"field_profile_contact_source";a:5:{s:6:"region";s:4:"main";s:6:"weight";s:1:"4";s:12:"has_required";b:0;s:5:"title";s:6:"Source";s:6:"hidden";i:0;}s:30:"field_profile_next_interaction";a:5:{s:6:"region";s:4:"main";s:6:"weight";s:1:"5";s:12:"has_required";b:0;s:5:"title";s:24:"Time of Next interaction";s:6:"hidden";i:0;}s:28:"field_profile_recent_webinar";a:5:{s:6:"region";s:4:"main";s:6:"weight";s:1:"6";s:12:"has_required";b:0;s:5:"title";s:28:"Recent Webinar Attended Date";s:6:"hidden";i:0;}s:27:"field_profile_organisms_yes";a:5:{s:6:"region";s:4:"main";s:6:"weight";s:1:"7";s:12:"has_required";b:0;s:5:"title";s:21:"Organisms of Interest";s:6:"hidden";i:0;}s:19:"field_profile_phone";a:5:{s:6:"region";s:4:"main";s:6:"weight";s:1:"1";s:12:"has_required";b:0;s:5:"title";s:15:"Secondary Phone";s:6:"hidden";i:0;}s:25:"field_profile_not_solicit";a:5:{s:6:"region";s:4:"main";s:6:"weight";s:1:"2";s:12:"has_required";b:0;s:5:"title";s:16:"Email Preference";s:6:"hidden";i:0;}s:20:"field_profile_domain";a:5:{s:6:"region";s:4:"main";s:6:"weight";s:1:"9";s:12:"has_required";b:0;s:5:"title";s:6:"Domain";s:6:"hidden";i:1;}s:6:"domain";a:5:{s:6:"region";s:6:"footer";s:6:"weight";s:1:"0";s:12:"has_required";b:1;s:5:"title";s:21:"Domain access options";s:9:"collapsed";i:1;}i:0;a:5:{s:6:"region";s:6:"footer";s:6:"weight";s:1:"1";s:12:"has_required";b:0;s:5:"title";N;s:6:"hidden";i:0;}s:10:"xmlsitemap";a:6:{s:6:"region";s:6:"footer";s:6:"weight";s:1:"3";s:12:"has_required";b:0;s:5:"title";s:11:"XML sitemap";s:9:"collapsed";i:1;s:6:"hidden";i:0;}s:9:"nodewords";a:6:{s:6:"region";s:6:"footer";s:6:"weight";s:1:"2";s:12:"has_required";b:0;s:5:"title";s:9:"Meta tags";s:9:"collapsed";i:1;s:6:"hidden";i:0;}}" string(9) "s:2:"30";" string(8) "s:1:"5";" string(9) "s:2:"20";" string(10) "s:3:"200";" string(8) "s:1:"7";" string(9) "s:2:"30";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(8) "s:1:"7";" string(15) "s:8:"mimemail";" string(22) "s:14:"uploaded_files";" string(8) "s:1:"7";" string(4) "i:0;" string(4) "i:1;" string(4) "i:1;" string(6) "a:0:{}" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(13) "i:1598159158;" string(28) "s:20:"varnita@strandls.com";" string(4) "i:0;" string(63) "s:55:"sites/all/modules/date/date_popup/themes/datepicker.css";" string(4) "i:1;" string(4) "i:0;" string(191) "s:182:"Type the below code ?This question is for testing whether you are a human visitor and to prevent automated spam submissions.
";" string(111) "s:102:"user-login-form user-register user-pass user-login webform-client-form-58 webform-client-form-248";" string(4) "i:0;" string(4) "i:1;" string(8) "s:1:"2";" string(7) "s:0:"";" string(4) "i:1;" string(8) "s:1:"0";" string(8) "s:1:"0";" string(11) "s:4:"fast";" string(10) "s:3:"800";" string(36) "s:28:"Containing none of the words";" string(20) "s:12:"nodecomments";" string(8) "s:1:"0";" string(4) "i:1;" string(8) "s:1:"0";" string(8) "s:1:"3";" string(4) "i:0;" string(4) "i:1;" string(4) "i:1;" string(4) "i:0;" string(4) "i:0;" string(4) "i:1;" string(4) "i:1;" string(52) "a:2:{s:6:"status";s:1:"0";s:8:"priority";s:3:"0.5";}" string(8) "s:1:"0";" string(4) "i:0;" string(21) "s:13:"post-register";" string(72) "s:64:"user/%uid/edit?destination=user/%uid/edit/Personal%20information";" string(4) "i:0;" string(8) "s:1:"0";" string(6) "i:217;" string(11) "s:4:"none";" string(8) "s:1:"0";" string(7) "s:0:"";" string(4) "i:1;" string(6) "a:0:{}" string(8) "s:1:"1";" string(8) "s:1:"1";" string(15) "s:8:"vertical";" string(23) "a:1:{i:0;s:6:"enable";}" string(15) "s:8:"comments";" string(6) "a:0:{}" string(11) "s:4:"type";" string(11) "s:4:"each";" string(13) "i:1598158809;" string(23) "a:1:{i:0;s:6:"status";}" string(7) "s:0:"";" string(8) "s:1:"1";" string(4) "i:1;" string(4) "i:0;" string(4) "i:1;" string(45) "s:37:"form-dfd30ee332d09b46662528000b81bac1";" string(15) "s:8:"disabled";" string(4) "i:0;" string(4) "i:0;" string(4) "i:1;" string(8) "s:1:"0";" string(4) "i:0;" string(8) "s:1:"1";" string(4) "i:1;" string(7) "s:0:"";" string(8) "s:1:"2";" string(8) "s:1:"0";" string(4) "i:0;" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(8) "s:1:"0";" string(4) "i:1;" string(8) "s:1:"0";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(25) "s:17:"faq/[catpath-raw]";" string(8) "s:1:"1";" string(7) "s:0:"";" string(7) "s:0:"";" string(37) "s:29:"tickets/[field_ticket_id-raw]";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(28) "s:20:"profiles/[title-raw]";" string(7) "s:0:"";" string(7) "s:0:"";" string(8) "s:1:"0";" string(7) "s:0:"";" string(6) "a:0:{}" string(11) "s:4:"each";" string(11) "s:4:"type";" string(6) "a:0:{}" string(8) "s:1:"1";" string(23) "a:1:{i:0;s:6:"enable";}" string(41) "a:2:{i:0;s:6:"status";i:1;s:7:"promote";}" string(8) "s:1:"0";" string(8) "s:1:"0";" string(4) "i:1;" string(4) "i:0;" string(4) "i:1;" string(45) "s:37:"form-cde039f445598285d1778ea06f2e80ea";" string(15) "s:8:"disabled";" string(4) "i:1;" string(8) "s:1:"1";" string(8) "s:1:"2";" string(4) "i:1;" string(6) "a:0:{}" string(7) "s:0:"";" string(4) "i:0;" string(8) "s:1:"0";" string(8) "s:1:"4";" string(8) "s:1:"1";" string(9) "s:2:"50";" string(8) "s:1:"3";" string(4) "i:0;" string(8) "s:1:"1";" string(8) "s:1:"1";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(11) "s:4:"none";" string(4) "i:0;" string(7) "s:0:"";" string(15) "s:8:"comments";" string(20) "s:12:"nodecomments";" string(7) "s:0:"";" string(8) "s:1:"0";" string(8) "s:1:"2";" string(10) "s:3:"768";" string(8) "s:1:"0";" string(7) "s:0:"";" string(8) "s:1:"0";" string(7) "s:0:"";" string(4) "i:0;" string(4) "i:0;" string(7) "s:0:"";" string(4) "i:0;" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(4) "i:1;" string(9) "s:2:"12";" string(9) "s:2:"27";" string(4) "i:0;" string(7) "s:0:"";" string(8) "s:1:"1";" string(8) "s:1:"1";" string(4) "i:1;" string(4) "i:1;" string(8) "s:1:"0";" string(10) "s:3:"768";" string(8) "s:1:"0";" string(4) "i:1;" string(13) "s:6:"always";" string(7) "s:0:"";" string(4) "i:1;" string(13) "s:6:"static";" string(11) "s:4:"user";" string(8) "s:1:"2";" string(6) "a:0:{}" string(7) "s:0:"";" string(4) "i:0;" string(8) "s:1:"0";" string(8) "s:1:"4";" string(8) "s:1:"1";" string(9) "s:2:"50";" string(4) "i:0;" string(23) "s:15:"faq/[title-raw]";" string(32) "s:24:"users/[user-raw]/contact";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(8) "s:1:"0";" string(4) "i:1;" string(8) "s:1:"0";" string(7) "s:0:"";" string(4) "i:0;" string(4) "i:1;" string(4) "i:1;" string(7) "s:0:"";" string(4) "i:1;" string(2) "N;" string(4) "i:0;" string(4) "i:0;" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(12) "s:5:"block";" string(12) "s:5:"block";" string(12) "s:5:"block";" string(12) "s:5:"block";" string(12) "s:5:"block";" string(8) "s:1:"0";" string(12) "s:5:"block";" string(12) "s:5:"block";" string(8) "s:1:"2";" string(12) "s:5:"block";" string(8) "s:1:"3";" string(12) "s:5:"block";" string(9) "s:2:"15";" string(10) "s:3:"128";" string(13) "s:6:"select";" string(4) "i:1;" string(29) "s:21:"Customize your search";" string(8) "s:1:"+";" string(12) "s:5:"block";" string(78) "s:70:"jpg jpeg gif png txt doc xls pdf ppt zip rar tar py docx xlsx pptx log";" string(8) "s:1:"2";" string(10) "s:3:"100";" string(78) "s:70:"jpg jpeg gif png txt doc xls pdf ppt zip rar tar py docx xlsx pptx log";" string(8) "s:1:"2";" string(10) "s:3:"100";" string(78) "s:70:"jpg jpeg gif png txt doc xls pdf ppt zip rar tar py docx xlsx pptx log";" string(8) "s:1:"2";" string(10) "s:3:"100";" string(78) "s:70:"jpg jpeg gif png txt doc xls pdf ppt zip rar tar py docx xlsx pptx log";" string(8) "s:1:"2";" string(10) "s:3:"100";" string(78) "s:70:"jpg jpeg gif png txt doc xls pdf ppt zip rar tar py docx xlsx pptx log";" string(8) "s:1:"2";" string(10) "s:3:"100";" string(78) "s:70:"jpg jpeg gif png txt doc xls pdf ppt zip rar tar py docx xlsx pptx log";" string(8) "s:1:"2";" string(10) "s:3:"100";" string(78) "s:70:"jpg jpeg gif png txt doc xls pdf ppt zip rar tar py docx xlsx pptx log";" string(8) "s:1:"2";" string(10) "s:3:"100";" string(78) "s:70:"jpg jpeg gif png txt doc xls pdf ppt zip rar tar py docx xlsx pptx log";" string(10) "s:3:"100";" string(78) "s:70:"jpg jpeg gif png txt doc xls pdf ppt zip rar tar py docx xlsx pptx log";" string(8) "s:1:"2";" string(10) "s:3:"100";" string(78) "s:70:"jpg jpeg gif png txt doc xls pdf ppt zip rar tar py docx xlsx pptx log";" string(8) "s:1:"2";" string(10) "s:3:"100";" string(8) "s:1:"2";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(4) "i:0;" string(603) "a:27:{s:11:"interaction";s:11:"interaction";s:13:"supportticket";s:13:"supportticket";s:7:"account";i:0;s:6:"biblio";i:0;s:7:"comment";i:0;s:5:"event";i:0;s:3:"faq";i:0;s:5:"image";i:0;s:8:"internal";i:0;s:19:"non_searchable_page";i:0;s:11:"opportunity";i:0;s:20:"support_organization";i:0;s:4:"page";i:0;s:7:"product";i:0;s:12:"product_page";i:0;s:18:"product_quote_form";i:0;s:7:"profile";i:0;s:6:"region";i:0;s:16:"software_product";i:0;s:5:"story";i:0;s:15:"support_product";i:0;s:6:"survey";i:0;s:5:"video";i:0;s:7:"webform";i:0;s:14:"webform_report";i:0;s:12:"testimonials";i:0;s:7:"webinar";i:0;}" string(12) "s:5:"_self";" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(7) "s:0:"";" string(15) "s:8:"existing";" string(11) "s:4:"1001";" string(4) "i:1;" string(4) "i:0;" string(13) "s:6:"ignore";" string(4) "i:0;" string(4) "b:0;" string(20) "s:12:"result_stats";" string(20) "s:12:"result_terms";" string(12) "s:5:"infos";" string(18) "s:10:"by_message";" string(4) "i:0;" string(8) "s:1:"1";" string(4) "i:0;" string(22) "s:14:"tree_structure";" string(4) "i:1;" string(11) "s:4:"file";" string(12) "s:5:"comma";" string(8) "s:1:"2";" string(7) "s:0:"";" string(11) "s:4:"none";" string(7) "s:0:"";" string(4) "i:0;" string(33) "s:25:"[page-title] | Strand NGS";" string(72) "s:64:"Strand NGS | Global Next generation sequencing analysis software";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(7) "s:0:"";" string(4) "i:0;" string(4) "i:0;" string(4) "i:0;" string(4) "i:0;" string(4) "i:1;" string(4) "i:0;" string(4) "i:0;" string(4) "i:0;" string(4) "i:0;" string(4) "i:0;" string(4) "i:0;" string(4) "i:0;" string(4) "i:0;" string(4) "i:0;" string(4) "i:0;" string(4) "i:0;" string(4) "i:0;" string(4) "i:0;" string(4) "i:0;" string(4) "i:1;" string(4) "i:0;" string(4) "i:0;" string(4) "i:0;" string(4) "i:0;" string(4) "i:0;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(4) "i:1;" string(15) "s:8:"disabled";" string(26) "s:18:"Filter the results";" string(4) "i:0;" string(4) "i:1;" string(4) "i:0;" string(4) "i:1;" string(4) "i:0;" string(4) "i:1;" string(4) "i:0;" string(7) "s:0:"";" string(4) "i:0;" string(4) "i:1;" string(4) "i:0;" string(4) "i:0;" string(4) "i:0;" string(4) "i:0;" string(4) "i:1;" string(4) "i:0;" string(4) "i:0;" string(4) "i:0;" string(4) "i:0;" string(4) "i:0;" string(7) "s:0:"";" string(22) "s:14:"tree_structure";" string(24) "a:1:{i:1001;s:4:"1001";}" string(12) "s:5:"comma";" string(7) "s:0:"";" string(16) "s:9:"quotation";" string(7) "s:0:"";" string(11) "s:4:"Unix";" string(11) "s:4:"name";" string(22) "s:14:"name_if_needed";" string(11) "s:4:"none";" string(4) "i:1;" string(7) "s:0:"";" string(7) "s:0:"";" string(11) "s:4:"both";" string(11) "s:4:"show";" string(7) "s:0:"";" string(7) "s:0:"";" string(11) "s:4:"both";" string(7) "s:0:"";" string(7) "s:0:"";" string(4) "i:0;" string(11) "s:4:"show";" string(7) "s:0:"";" string(12) "s:5:"4.9.1";" string(7) "s:0:"";" string(11) "s:4:"both";" string(159) "s:150:"http://apps.isiknowledge.com/InboundService.do?Func=Frame&product=WOS&action=retrieve&SrcApp=EndNote&Init=Yes&SrcAuth=ResearchSoft&mode=FullRecord&UT=";" string(13) "s:6:"biblio";" string(13) "s:6:"Biblio";" string(9) "s:2:"25";" string(4) "i:0;" string(4) "i:0;" string(12) "s:5:"86400";" string(8) "s:1:"4";" string(4) "i:0;" string(8) "s:1:"4";" string(16) "s:9:"n.created";" string(4) "i:0;" string(4) "i:1;" string(7) "s:0:"";" string(10) "s:3:"nid";" string(10) "s:3:"nid";" string(7) "s:0:"";" string(4) "i:0;" string(7) "s:0:"";" string(4) "i:0;" string(4) "i:0;" string(8) "s:1:"0";" string(98) "a:4:{s:3:"rtf";s:3:"rtf";s:6:"tagged";s:6:"tagged";s:3:"xml";s:3:"xml";s:6:"bibtex";s:6:"bibtex";}" string(4) "i:1;" string(4) "i:0;" string(4) "i:0;" string(4) "i:0;" string(4) "i:0;" string(4) "i:1;" string(4) "i:0;" string(4) "i:0;" string(7) "s:0:"";" string(18) "s:10:"Biblio:CRM";" string(7) "s:0:"";" string(4) "i:0;" string(4) "i:0;" string(4) "i:0;" string(4) "i:0;" string(21) "s:13:"Biblio search";" string(4) "i:0;" string(11) "s:4:"year";" string(118) "a:5:{s:6:"author";s:6:"author";s:5:"title";s:5:"title";s:4:"type";s:4:"type";s:4:"year";s:4:"year";s:7:"keyword";i:0;}" string(4) "i:0;" string(11) "s:4:"DESC";" string(16) "s:9:"Submitted";" string(15) "s:8:"In Press";" string(10) "s:3:"cse";" string(14) "s:7:"tabular";" string(11) "s:4:"none";" string(4) "i:0;" string(9) "s:2:"10";" string(8) "s:1:"1";" string(614) "a:3:{s:32:"c31b0fe3cc9e58e9868672fc9882f325";O:8:"stdClass":3:{s:8:"filename";s:66:"sites/all/modules/captcha/image_captcha/fonts/Tuffy/Tuffy_Bold.ttf";s:8:"basename";s:14:"Tuffy_Bold.ttf";s:4:"name";s:10:"Tuffy_Bold";}s:32:"ce92a4de20aa1d6efd138e3cd8dfe600";O:8:"stdClass":3:{s:8:"filename";s:61:"sites/all/modules/captcha/image_captcha/fonts/Tuffy/Tuffy.ttf";s:8:"basename";s:9:"Tuffy.ttf";s:4:"name";s:5:"Tuffy";}s:32:"2d0e63850e91ee2c999b22e41eb0b20c";O:8:"stdClass":3:{s:8:"filename";s:61:"sites/all/modules/captcha/image_captcha/fonts/Tesox/tesox.ttf";s:8:"basename";s:9:"tesox.ttf";s:4:"name";s:5:"tesox";}}" string(1832) "a:41:{i:0;s:20:"event_comment_insert";i:1;s:20:"event_comment_update";i:2;s:20:"event_comment_delete";i:3;s:18:"event_comment_view";i:4;s:21:"event_comment_publish";i:5;s:23:"event_comment_unpublish";i:9;s:15:"event_node_view";i:10;s:17:"event_node_delete";i:11;s:10:"event_init";i:13;s:26:"event_taxonomy_term_insert";i:14;s:26:"event_taxonomy_term_update";i:16;s:17:"event_user_update";i:17;s:15:"event_user_view";i:18;s:17:"event_user_delete";i:19;s:16:"event_user_login";i:20;s:17:"event_user_logout";i:21;s:39:"event_flag_flagged_ignore_flag_followup";i:22;s:41:"event_flag_unflagged_ignore_flag_followup";i:23;s:46:"event_flag_flagged_ignore_flag_interactiontime";i:24;s:48:"event_flag_unflagged_ignore_flag_interactiontime";i:25;s:50:"event_flag_flagged_ignore_flag_substansiveresponse";i:26;s:52:"event_flag_unflagged_ignore_flag_substansiveresponse";i:27;s:37:"event_flag_flagged_knowledge_base_ngs";i:28;s:39:"event_flag_unflagged_knowledge_base_ngs";i:29;s:41:"event_flag_flagged_contact_user_quarterly";i:30;s:43:"event_flag_unflagged_contact_user_quarterly";i:31;s:48:"event_flag_flagged_ignore_support_engineers_flag";i:32;s:50:"event_flag_unflagged_ignore_support_engineers_flag";i:33;s:44:"event_flag_flagged_flag_substansive_response";i:34;s:46:"event_flag_unflagged_flag_substansive_response";i:35;s:34:"event_flag_flagged_user_to_contact";i:36;s:36:"event_flag_unflagged_user_to_contact";i:37;s:41:"event_flag_flagged_flag_support_engineers";i:38;s:43:"event_flag_unflagged_flag_support_engineers";i:39;s:43:"event_flag_flagged_flag_ticket_closure_rate";i:40;s:45:"event_flag_unflagged_flag_ticket_closure_rate";i:41;s:32:"event_flag_flagged_flag_followup";i:42;s:34:"event_flag_unflagged_flag_followup";i:43;s:28:"event_flag_flagged_bookmarks";i:44;s:30:"event_flag_unflagged_bookmarks";i:55;s:11:"rules_set_1";}"Strand NGS supports an extensive workflow for the analysis and visualization of Methyl-Seq data – such as from whole genome or targeted experiments. The workflow includes the ability to detect methylation in individual samples which can be used to further look at differentially methylated cytosines across samples/target regions and also study methylation effects at the genic level. Further downstream analysis such as GO, pathway analysis, etc can be performed on the set of affected genes.
Download the Methyl-Seq Highlights Guide Watch the Methyl-Seq Webinar Recording
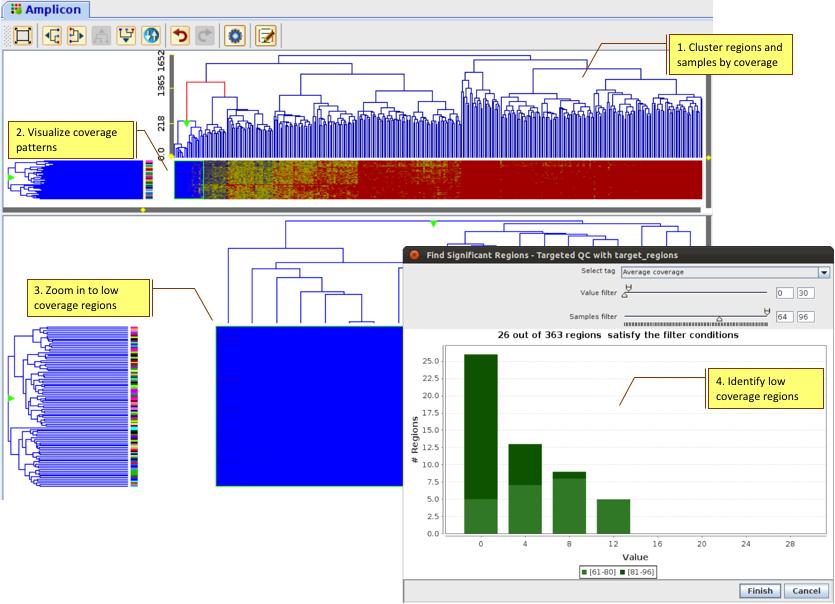
Evaluate efficacy of targeted re-sequencing, and identify regions with low coverage across samples.
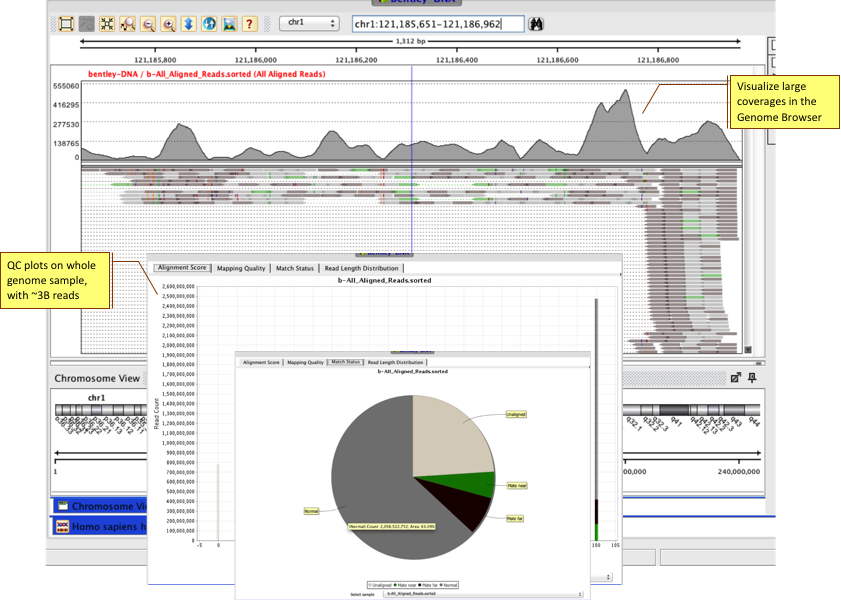
Perform Whole Genome analysis on human or other organisms on your desktop.
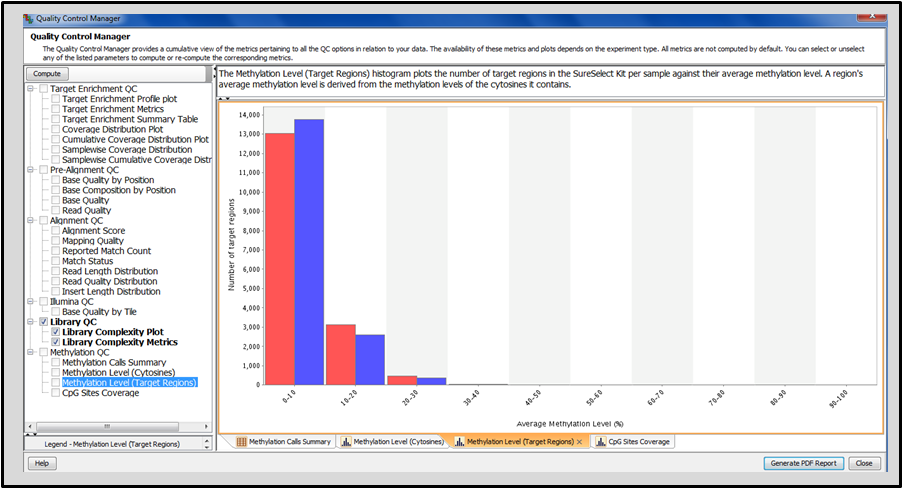
Use the QC manager to look at quality metrics for pre and post alignment, target enrichment and library complexity. Compute methylation specific QC to get information on methylation levels and CpG sites coverage for your samples.
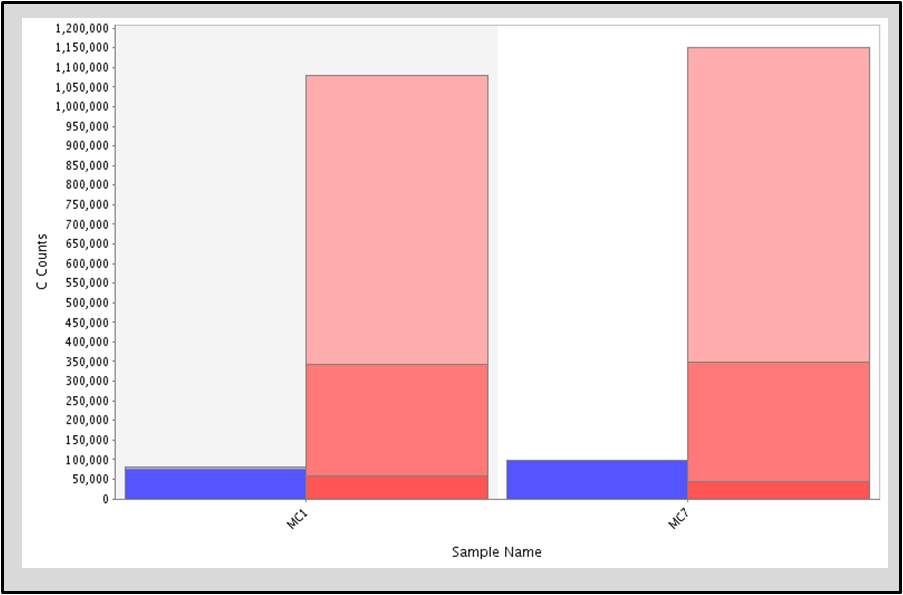
Identify methylated cytosines for specific loci on the genome for individual samples. Algorithm uses a variety of parameters such as bisulfite conversion error rate, sequencing error, read coverage, base quality and methylation fraction cut-offs for heightened accuracy.
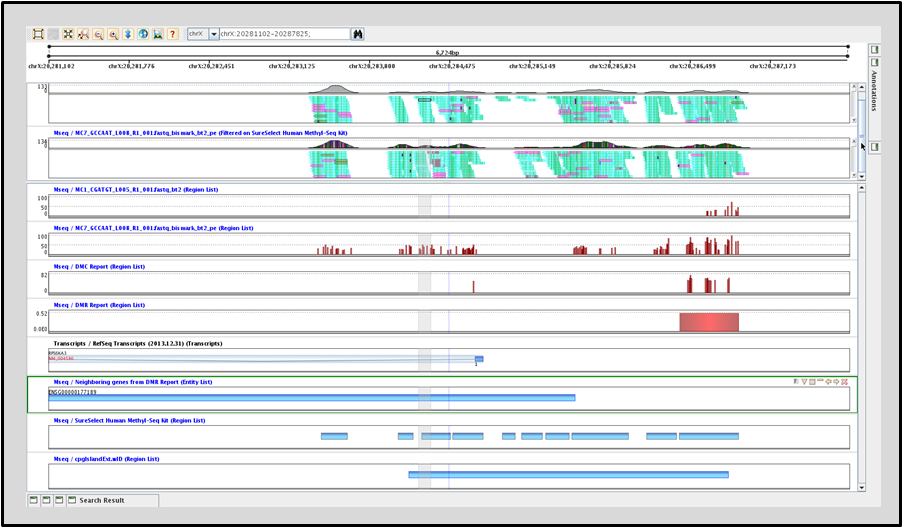
Identify differentially methylated cytosines (DMCs) across experimental conditions. Use this information to further discover differentially methylated regions across these conditions.
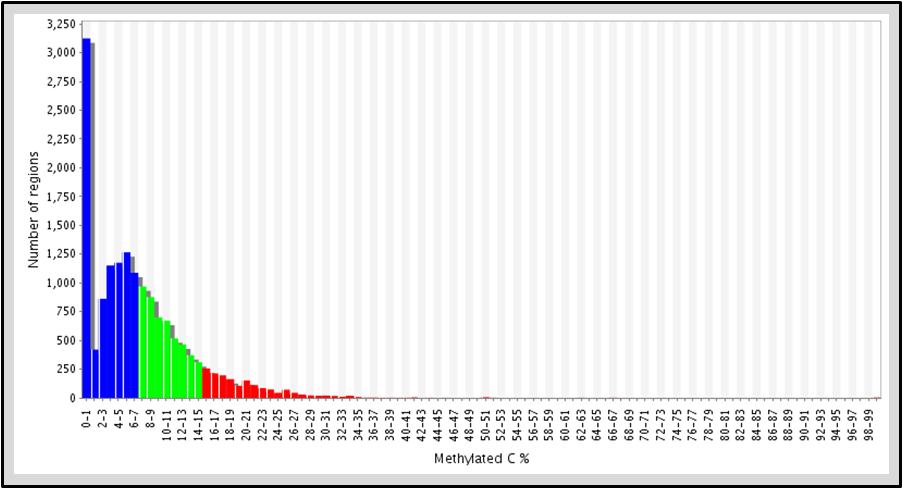
Identify regions with low, moderate, and high methylation density within an individual sample for target regions of interest.
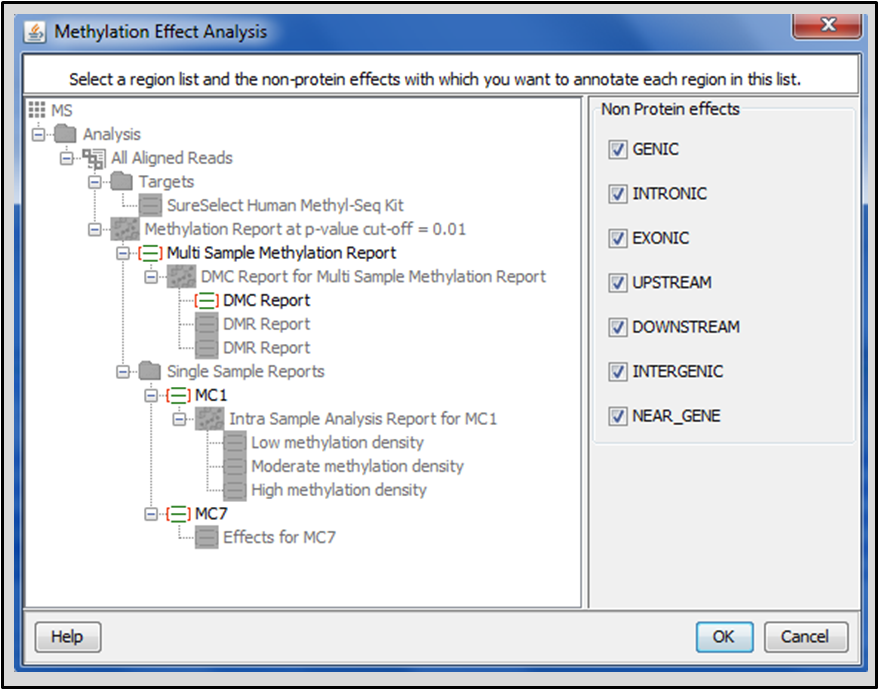
Perform Methylation Effect Analysis to determine the genomic context of the methylated or differentially methylated cytosines resulting from Methylation Detection and DMC analyses. Strand NGS can annotate these cytosines with the selected non-protein effects.
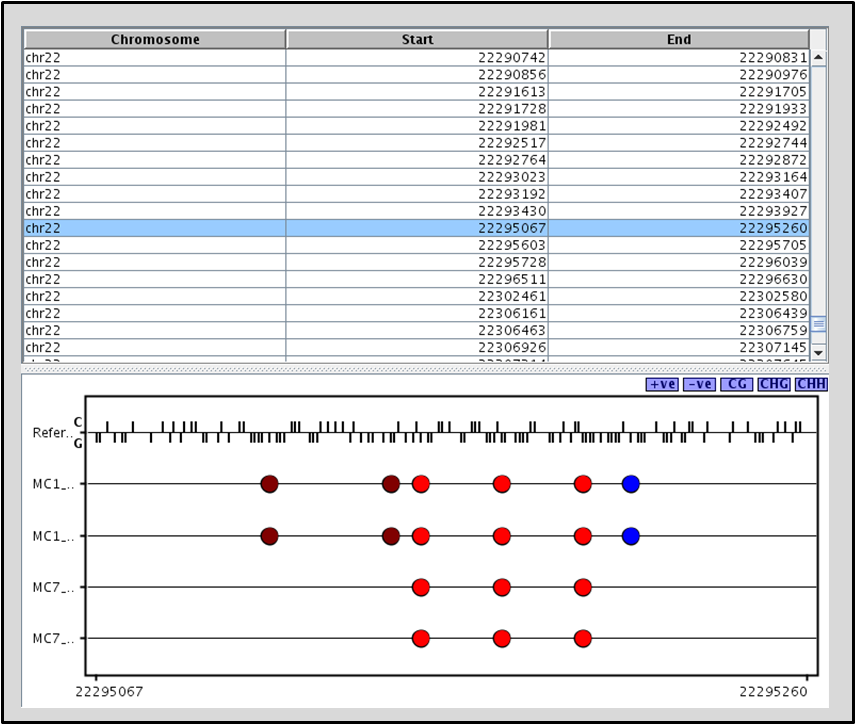
Use the Lollipop Plot to visualize methylated or differentially methylated cytosines by regions. The interactive genome browser is customized for displaying bisulfite converted reads. Within the GB, the methylation level histogram helps visualize the proportion of methylated cytosines compared to unmethylated cytosines in the read coverage.
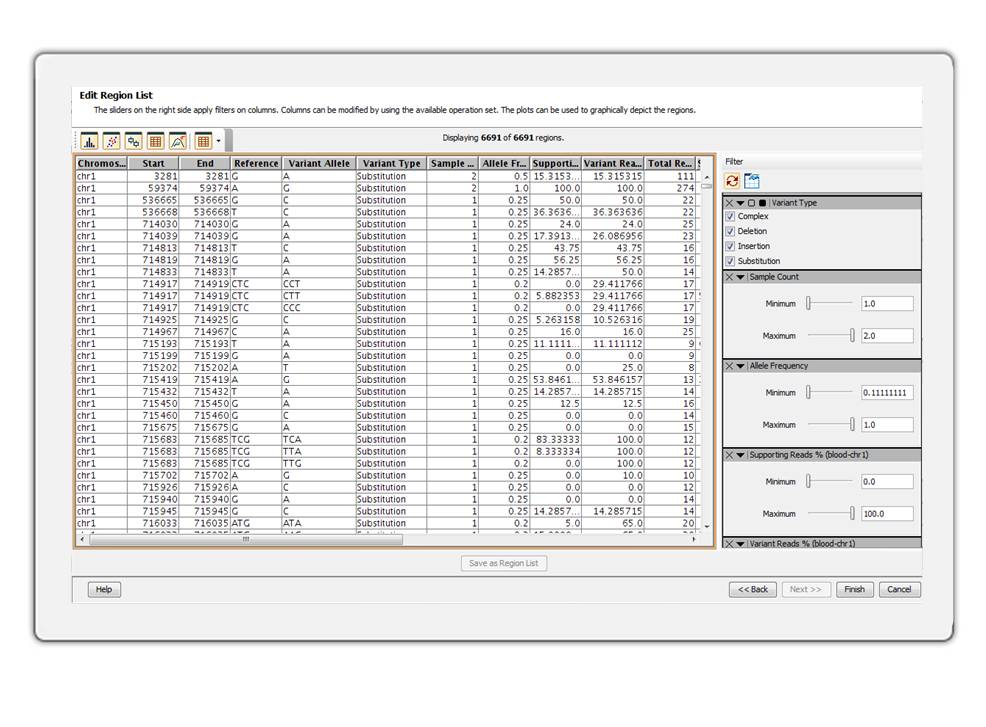
Seamlessly create and manipulate genomic region lists. Filter region lists based on different attributes.
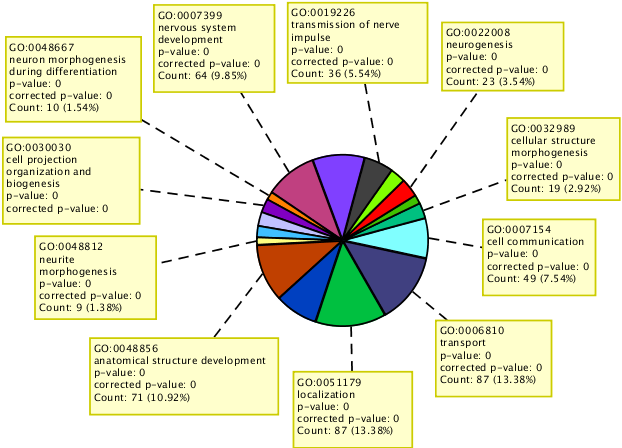
Perform GO analysis on the set of genes affected by identified variants.
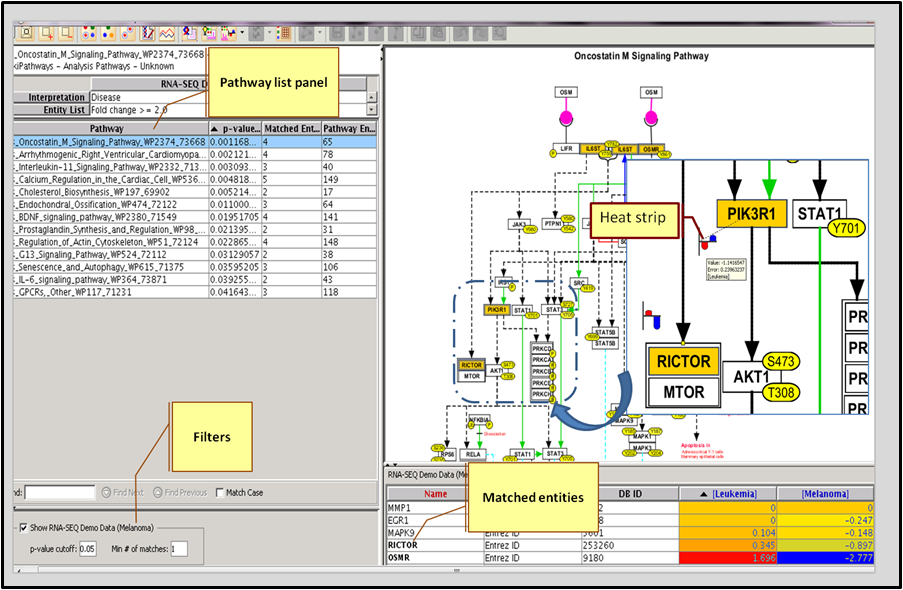
Use the packaged database of 2 million interactions (with supporting PubMed references) to find relationships between genes. Learn more
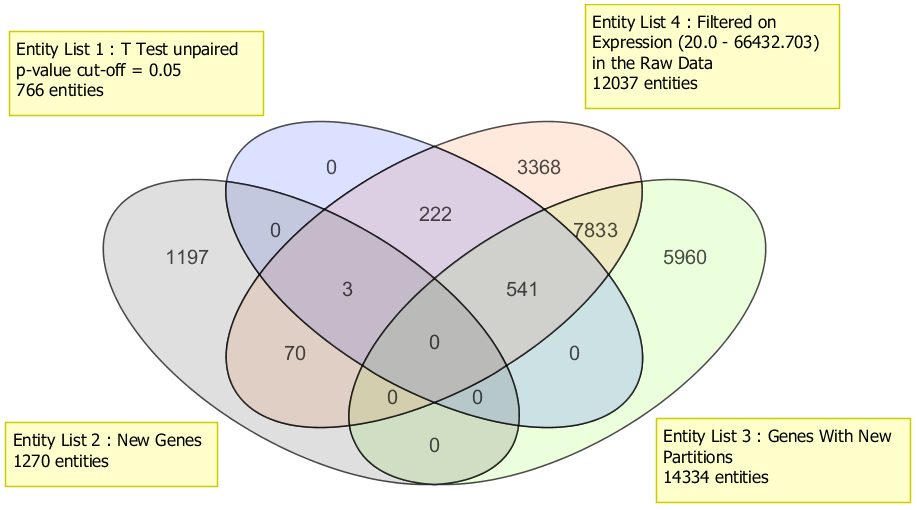
Compare different gene lists from multiple experiments and across organisms.
2018 © Strand Life Sciences Pvt Ltd. All rights reserved.